当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Estimation of Drug-Target Residence Time by Targeted Molecular Dynamics Simulations
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-11-09 , DOI: 10.1021/acs.jcim.2c00852 Sonia Ziada 1 , Julien Diharce 1 , Eric Raimbaud 2 , Samia Aci-Sèche 1 , Pierre Ducrot 2 , Pascal Bonnet 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-11-09 , DOI: 10.1021/acs.jcim.2c00852 Sonia Ziada 1 , Julien Diharce 1 , Eric Raimbaud 2 , Samia Aci-Sèche 1 , Pierre Ducrot 2 , Pascal Bonnet 1
Affiliation
|
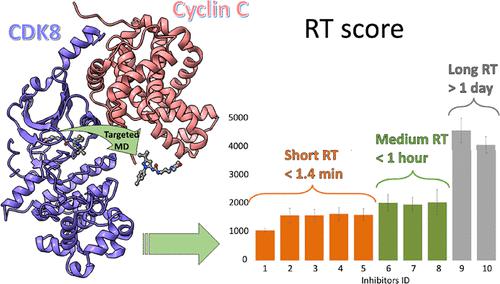
Drug-target residence time has emerged as a key selection factor in drug discovery since the binding duration of a drug molecule to its protein target can significantly impact its in vivo efficacy. The challenge in studying the residence time, in early drug discovery stages, lies in how to cost-effectively determine the residence time for the systematic assessment of compounds. Currently, there is still a lack of computational protocols to quickly estimate such a measure, particularly for large and flexible protein targets and drugs. Here, we report an efficient computational protocol, based on targeted molecular dynamics, to rank drug candidates by their residence time and to obtain insights into ligand–target dissociation mechanisms. The method was assessed on a dataset of 10 arylpyrazole inhibitors of CDK8, a large, flexible, and clinically important target, for which the experimental residence time of the inhibitors ranges from minutes to hours. The compounds were correctly ranked according to their estimated residence time scores compared to their experimental values. The analysis of protein–ligand interactions along the dissociation trajectories highlighted the favorable contribution of hydrophobic contacts to residence time and revealed key residues that strongly affect compound residence time.
中文翻译:

通过靶向分子动力学模拟估算药物靶标停留时间
药物靶标停留时间已成为药物发现的关键选择因素,因为药物分子与其蛋白质靶标的结合持续时间会显着影响其在体内的作用功效。在早期药物发现阶段研究停留时间的挑战在于如何经济高效地确定停留时间以对化合物进行系统评估。目前,仍然缺乏计算协议来快速估计这种措施,特别是对于大型和灵活的蛋白质靶标和药物。在这里,我们报告了一种基于靶向分子动力学的有效计算方案,根据候选药物的停留时间对候选药物进行排名,并深入了解配体-靶标解离机制。该方法在 CDK8 的 10 种芳基吡唑抑制剂的数据集上进行了评估,CDK8 是一个大的、灵活的、临床上重要的目标,抑制剂的实验停留时间从几分钟到几小时不等。根据与其实验值相比的估计停留时间得分,化合物被正确排序。沿着解离轨迹对蛋白质-配体相互作用的分析突出了疏水接触对停留时间的有利贡献,并揭示了强烈影响化合物停留时间的关键残基。
更新日期:2022-11-09
中文翻译:
通过靶向分子动力学模拟估算药物靶标停留时间
药物靶标停留时间已成为药物发现的关键选择因素,因为药物分子与其蛋白质靶标的结合持续时间会显着影响其在体内的作用功效。在早期药物发现阶段研究停留时间的挑战在于如何经济高效地确定停留时间以对化合物进行系统评估。目前,仍然缺乏计算协议来快速估计这种措施,特别是对于大型和灵活的蛋白质靶标和药物。在这里,我们报告了一种基于靶向分子动力学的有效计算方案,根据候选药物的停留时间对候选药物进行排名,并深入了解配体-靶标解离机制。该方法在 CDK8 的 10 种芳基吡唑抑制剂的数据集上进行了评估,CDK8 是一个大的、灵活的、临床上重要的目标,抑制剂的实验停留时间从几分钟到几小时不等。根据与其实验值相比的估计停留时间得分,化合物被正确排序。沿着解离轨迹对蛋白质-配体相互作用的分析突出了疏水接触对停留时间的有利贡献,并揭示了强烈影响化合物停留时间的关键残基。


























 京公网安备 11010802027423号
京公网安备 11010802027423号