当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Explainable Solvation Free Energy Prediction Combining Graph Neural Networks with Chemical Intuition
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-11-01 , DOI: 10.1021/acs.jcim.2c01013 Kaycee Low 1 , Michelle L Coote 2 , Ekaterina I Izgorodina 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-11-01 , DOI: 10.1021/acs.jcim.2c01013 Kaycee Low 1 , Michelle L Coote 2 , Ekaterina I Izgorodina 1
Affiliation
|
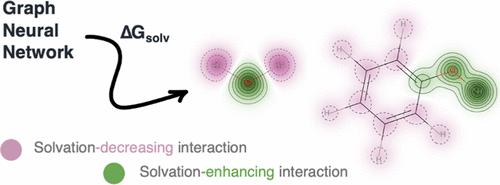
The prediction of a molecule’s solvation Gibbs free (ΔGsolv) energy in a given solvent is an important task which has traditionally been carried out via quantum chemical continuum methods or force field-based molecular simulations. Machine learning (ML) and graph neural networks in particular have emerged as powerful techniques for elucidating structure–property relationships. This work presents a graph neural network (GNN) for the prediction of ΔGsolv which, in addition to encoding typical atom and bond-level features, incorporates chemically intuitive, solvation-relevant parameters into the featurization process: semiempirical partial atomic charges and solvent dielectric constant. Solute–solvent interactions are included via an interaction map layer which can be visualized to examine solubility-enhancing or -decreasing interactions learnt by the model. On a test set of small organic molecules, our GNN predicts ΔGsolv in water and cyclohexane with an accuracy comparable to polarizable and ab initio generated force field methods [mean absolute error (MAE) = 0.4 and 0.2 kcal mol–1, respectively], without the need for any molecular simulation. For the FreeSolv data set of hydration free energies, the test MAE is 0.7 kcal mol–1. Interpretability and applicability of the model is highlighted through several examples including rationalizing the increased solubility of modified diaminoanthraquinones in organic solvents. The clear explanations afforded by our GNN allow for easy understanding of the model’s predictions, giving the experimental chemist confidence in employing ML models toward more optimized synthetic routes.
中文翻译:

结合图神经网络和化学直觉的可解释溶剂化自由能预测
预测给定溶剂中分子的溶剂化吉布斯自由 (Δ G solv ) 能量是一项重要任务,传统上通过量子化学连续介质方法或基于力场的分子模拟来执行。机器学习 (ML),尤其是图神经网络已成为阐明结构-性质关系的强大技术。这项工作提出了一个图神经网络 (GNN) 用于预测 Δ G solv其中,除了对典型的原子和键级特征进行编码外,还将化学上直观的、溶剂化相关的参数纳入特征化过程:半经验部分原子电荷和溶剂介电常数。溶质-溶剂相互作用通过相互作用图层包含在内,可以将其可视化以检查模型学习的溶解度增强或溶解度降低的相互作用。在小有机分子的测试集上,我们的 GNN 预测 Δ G溶解在水和环己烷中,其精度与可极化和从头算产生的力场方法相当 [平均绝对误差 (MAE) = 0.4 和 0.2 kcal mol –1,分别],而不需要任何分子模拟。对于水合自由能的 FreeSolv 数据集,测试 MAE 为 0.7 kcal mol –1。该模型的可解释性和适用性通过几个例子得到强调,包括合理化改性二氨基蒽醌在有机溶剂中溶解度的增加。我们的 GNN 提供的清晰解释可以让您轻松理解模型的预测,从而让实验化学家有信心将 ML 模型用于更优化的合成路线。
更新日期:2022-11-01
中文翻译:
结合图神经网络和化学直觉的可解释溶剂化自由能预测
预测给定溶剂中分子的溶剂化吉布斯自由 (Δ G solv ) 能量是一项重要任务,传统上通过量子化学连续介质方法或基于力场的分子模拟来执行。机器学习 (ML),尤其是图神经网络已成为阐明结构-性质关系的强大技术。这项工作提出了一个图神经网络 (GNN) 用于预测 Δ G solv其中,除了对典型的原子和键级特征进行编码外,还将化学上直观的、溶剂化相关的参数纳入特征化过程:半经验部分原子电荷和溶剂介电常数。溶质-溶剂相互作用通过相互作用图层包含在内,可以将其可视化以检查模型学习的溶解度增强或溶解度降低的相互作用。在小有机分子的测试集上,我们的 GNN 预测 Δ G溶解在水和环己烷中,其精度与可极化和从头算产生的力场方法相当 [平均绝对误差 (MAE) = 0.4 和 0.2 kcal mol –1,分别],而不需要任何分子模拟。对于水合自由能的 FreeSolv 数据集,测试 MAE 为 0.7 kcal mol –1。该模型的可解释性和适用性通过几个例子得到强调,包括合理化改性二氨基蒽醌在有机溶剂中溶解度的增加。我们的 GNN 提供的清晰解释可以让您轻松理解模型的预测,从而让实验化学家有信心将 ML 模型用于更优化的合成路线。

























 京公网安备 11010802027423号
京公网安备 11010802027423号