当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Density functional theory studies on molecular geometry, spectroscopy, HOMO–LUMO and reactivity descriptors of titanium(IV) and oxidozirconium(IV) complexes of phenylacetohydroxamic acid
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2022-09-27 , DOI: 10.1002/jcc.27004 Vineet Kumar Choudhary 1 , Kanika Mandhan 1 , Dibyajit Dash 2 , Sachin Bhardwaj 3 , Meena Kumari 3 , Neeraj Sharma 3
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2022-09-27 , DOI: 10.1002/jcc.27004 Vineet Kumar Choudhary 1 , Kanika Mandhan 1 , Dibyajit Dash 2 , Sachin Bhardwaj 3 , Meena Kumari 3 , Neeraj Sharma 3
Affiliation
|
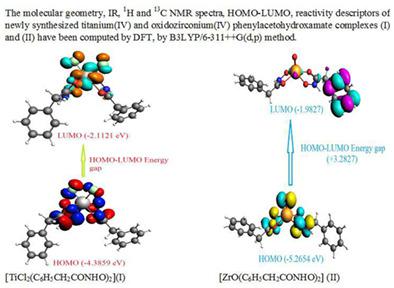
The molecular geometry of new titanium(IV) and oxidozirconium(IV) phenylacetohydroxamate complexes [TiCl2(L1)2] (I) and [ZrO(L1)2] (II) (where L1 = Potassium phenylacetohydroxamate = C6H5CH2CONHOK) computed by B3LYP/6-311++G(d,p) method has shown these to be distorted octahedral and square pyramidal, respectively. A comparison of computed characteristic bond lengths (CO, CN, and NO) of complexes with that of free ligand has shown chelation through carbonyl and hydroxamic oxygen atoms (O, O coordination). The TiO/ZrO bond lengths in complexes are suggestive of weak coordination through (carbonyl CO) and strong covalent (hydroxamic NO) bonding of the ligand. The magnitude of ClTiCl bond angle involving two chloride atoms is suggestive of cis-conformation at titanium metal in (I). The thermodynamic parameters Gibbs free energy, enthalpy, entropy, nuclear internal energy, constant volume heat capacity, and internal energy of ligand and complexes have been computed. From the energies of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), the global reactivity descriptors such as ionization potential (IP), electron affinity (EA), chemical potential (μ), hardness (η), softness (S), electronegativity (χ), electrophilicity index (ω), and dipole moment have been calculated. The computed vibrational frequencies, 1H and 13C NMR spectra have substantiated the molecular structure of complexes. The thermal behavior of complexes has been studied by thermogravimetric techniques (TGA, DTG, and DTA) in N2 atmosphere has shown complexes are thermally stable.
中文翻译:

苯乙酰异羟肟酸钛(IV)和氧化锆(IV)配合物的分子几何、光谱、HOMO-LUMO和反应性描述符的密度泛函理论研究
新型钛 (IV) 和氧化锆 (IV) 苯乙酰异羟肟酸盐配合物 [TiCl 2 (L1) 2 ] (I) 和 [ZrO(L1) 2 ] (II) 的分子几何结构(其中 L1 = 苯乙酰异羟肟酸钾 = C 6 H 5 CH 2 CONHOK) 通过 B3LYP/6-311++G(d,p) 方法计算表明它们分别是扭曲的八面体和方形金字塔。比较计算出的配合物与自由配体的特征键长(CO、CN 和 NO)表明通过羰基和异羟肟氧原子(O、O 配位)螯合。Ti O/Zr 配合物中的 O 键长表明通过配体的(羰基 C = O)弱配位和强共价键(异羟肟酸 NO)。涉及两个氯原子的 Cl Ti Cl 键角的大小暗示了 (I) 中钛金属的顺式构象。计算了热力学参数吉布斯自由能、焓、熵、核内能、定容热容以及配体和配合物的内能。从最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的能量,电离势(IP)、电子亲和力(EA)、化学势(μ)、硬度(η)、柔软度等全局反应性描述符(S )、电负性 ( χ )、亲电性指数 ( ω ) 和偶极矩已被计算。计算出的振动频率、1 H 和13 C NMR 谱证实了配合物的分子结构。在 N 2气氛中通过热重分析技术(TGA、DTG 和 DTA)研究了配合物的热行为,表明配合物是热稳定的。
更新日期:2022-09-27
中文翻译:
苯乙酰异羟肟酸钛(IV)和氧化锆(IV)配合物的分子几何、光谱、HOMO-LUMO和反应性描述符的密度泛函理论研究
新型钛 (IV) 和氧化锆 (IV) 苯乙酰异羟肟酸盐配合物 [TiCl 2 (L1) 2 ] (I) 和 [ZrO(L1) 2 ] (II) 的分子几何结构(其中 L1 = 苯乙酰异羟肟酸钾 = C 6 H 5 CH 2 CONHOK) 通过 B3LYP/6-311++G(d,p) 方法计算表明它们分别是扭曲的八面体和方形金字塔。比较计算出的配合物与自由配体的特征键长(CO、CN 和 NO)表明通过羰基和异羟肟氧原子(O、O 配位)螯合。Ti O/Zr 配合物中的 O 键长表明通过配体的(羰基 C = O)弱配位和强共价键(异羟肟酸 NO)。涉及两个氯原子的 Cl Ti Cl 键角的大小暗示了 (I) 中钛金属的顺式构象。计算了热力学参数吉布斯自由能、焓、熵、核内能、定容热容以及配体和配合物的内能。从最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的能量,电离势(IP)、电子亲和力(EA)、化学势(μ)、硬度(η)、柔软度等全局反应性描述符(S )、电负性 ( χ )、亲电性指数 ( ω ) 和偶极矩已被计算。计算出的振动频率、1 H 和13 C NMR 谱证实了配合物的分子结构。在 N 2气氛中通过热重分析技术(TGA、DTG 和 DTA)研究了配合物的热行为,表明配合物是热稳定的。

























 京公网安备 11010802027423号
京公网安备 11010802027423号