Complex & Intelligent Systems ( IF 5.8 ) Pub Date : 2022-08-23 , DOI: 10.1007/s40747-022-00846-y Machbah Uddin 1, 2 , Mohammad Khairul Islam 1 , Md Rakib Hassan 2 , Farah Jahan 1 , Joong Hwan Baek 3
|
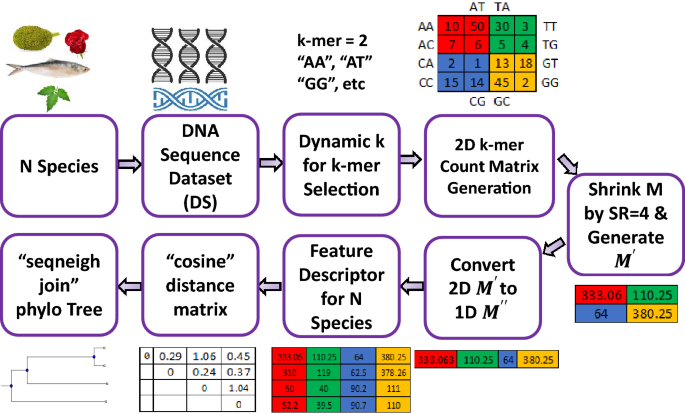
DNA sequence similarity analysis is necessary for enormous purposes including genome analysis, extracting biological information, finding the evolutionary relationship of species. There are two types of sequence analysis which are alignment-based (AB) and alignment-free (AF). AB is effective for small homologous sequences but becomes NP-hard problem for long sequences. However, AF algorithms can solve the major limitations of AB. But most of the existing AF methods show high time complexity and memory consumption, less precision, and less performance on benchmark datasets. To minimize these limitations, we develop an AF algorithm using a 2D \(k-mer\) count matrix inspired by the CGR approach. Then we shrink the matrix by analyzing the neighbors and then measure similarities using the best combinations of pairwise distance (PD) and phylogenetic tree methods. We also dynamically choose the value of k for \(k-mer\). We develop an efficient system for finding the positions of \(k-mer\) in the count matrix. We apply our system in six different datasets. We achieve the top rank for two benchmark datasets from AFproject, 100% accuracy for two datasets (16 S Ribosomal, 18 Eutherian), and achieve a milestone for time complexity and memory consumption in comparison to the existing study datasets (HEV, HIV-1). Therefore, the comparative results of the benchmark datasets and existing studies demonstrate that our method is highly effective, efficient, and accurate. Thus, our method can be used with the top level of authenticity for DNA sequence similarity measurement.
中文翻译:
一种快速高效的DNA序列相似性识别算法
DNA 序列相似性分析对于包括基因组分析、提取生物信息、发现物种进化关系在内的许多目的都是必要的。有两种类型的序列分析,即基于比对(AB)和无比对(AF)。AB 对于小的同源序列是有效的,但对于长序列就变成了NP -hard 问题。然而,AF 算法可以解决 AB 的主要局限性。但是大多数现有的 AF 方法在基准数据集上表现出高时间复杂度和内存消耗、精度较低和性能较低。为了最大限度地减少这些限制,我们开发了一种使用 2D \(k-mer\)受 CGR 方法启发的计数矩阵。然后我们通过分析邻居来缩小矩阵,然后使用成对距离 (PD) 和系统发育树方法的最佳组合来测量相似性。我们还为\(k-mer\)动态选择k的值。我们开发了一个有效的系统来寻找\(k-mer\)的位置在计数矩阵中。我们将我们的系统应用于六个不同的数据集。我们在 AFproject 的两个基准数据集上取得了最高排名,两个数据集(16 S Ribosomal,18 Eutherian)的准确率达到了 100%,并且与现有研究数据集(HEV,HIV-1)相比,时间复杂度和内存消耗达到了里程碑). 因此,基准数据集和现有研究的比较结果表明我们的方法是高效、高效和准确的。因此,我们的方法可以用于 DNA 序列相似性测量的最高真实性。
























 京公网安备 11010802027423号
京公网安备 11010802027423号