American Journal of Human Genetics ( IF 9.8 ) Pub Date : 2022-07-26 , DOI: 10.1016/j.ajhg.2022.06.013 Hugo Lemoine 1 , Loann Raud 1 , François Foulquier 2 , John A Sayer 3 , Baptiste Lambert 2 , Eric Olinger 4 , Siriane Lefèvre 5 , Bertrand Knebelmann 6 , Peter C Harris 7 , Pascal Trouvé 1 , Aurore Desprès 8 , Gabrielle Duneau 9 , Marie Matignon 10 , Anais Poyet 11 , Noémie Jourde-Chiche 12 , Dominique Guerrot 13 , Sandrine Lemoine 14 , Guillaume Seret 15 , Miguel Barroso-Gil 4 , Coralie Bingham 16 , Rodney Gilbert 17 , , , Yannick Le Meur 18 , Marie-Pierre Audrézet 19 , Emilie Cornec-Le Gall 20
|
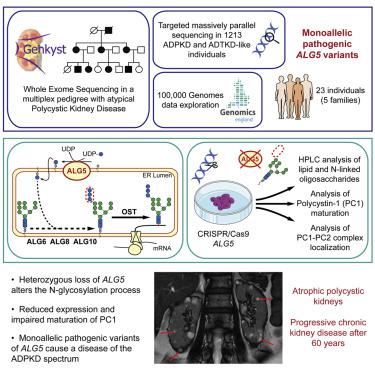
Disorders of the autosomal dominant polycystic kidney disease (ADPKD) spectrum are characterized by the development of kidney cysts and progressive kidney function decline. PKD1 and PKD2, encoding polycystin (PC)1 and 2, are the two major genes associated with ADPKD; other genes include IFT140, GANAB, DNAJB11, and ALG9. Genetic testing remains inconclusive in ∼7% of the families. We performed whole-exome sequencing in a large multiplex genetically unresolved (GUR) family affected by ADPKD-like symptoms and identified a monoallelic frameshift variant (c.703_704delCA) in ALG5. ALG5 encodes an endoplasmic-reticulum-resident enzyme required for addition of glucose molecules to the assembling N-glycan precursors. To identify additional families, we screened a cohort of 1,213 families with ADPKD-like and/or autosomal-dominant tubulointerstitial kidney diseases (ADTKD), GUR (n = 137) or naive to genetic testing (n = 1,076), by targeted massively parallel sequencing, and we accessed Genomics England 100,000 Genomes Project data. Four additional families with pathogenic variants in ALG5 were identified. Clinical presentation was consistent in the 23 affected members, with non-enlarged cystic kidneys and few or no liver cysts; 8 subjects reached end-stage kidney disease from 62 to 91 years of age. We demonstrate that ALG5 haploinsufficiency is sufficient to alter the synthesis of the N-glycan chain in renal epithelial cells. We also show that ALG5 is required for PC1 maturation and membrane and ciliary localization and that heterozygous loss of ALG5 affects PC1 maturation. Overall, our results indicate that monoallelic variants of ALG5 lead to a disorder of the ADPKD-spectrum characterized by multiple small kidney cysts, progressive interstitial fibrosis, and kidney function decline.
中文翻译:
单等位基因致病性 ALG5 变异导致非典型多囊肾病和间质纤维化
常染色体显性多囊肾病(ADPKD)谱系疾病的特征是肾囊肿的发展和进行性肾功能衰退。PKD1和PKD2编码多囊蛋白 (PC)1 和 2,是与 ADPKD 相关的两个主要基因;其他基因包括IFT140、GANAB、DNAJB11和ALG9。约 7% 的家庭的基因检测仍不确定。我们对受 ADPKD 样症状影响的大型多重遗传未解决 (GUR) 家族进行了全外显子组测序,并在 ALG5 中发现了单等位基因移码变异 (c.703_704delCA) 。ALG5编码一种内质网驻留酶,该酶是将葡萄糖分子添加到组装 N-聚糖前体中所需的。为了确定更多家庭,我们通过有针对性的大规模平行筛查,筛选了 1,213 个患有 ADPKD 样和/或常染色体显性肾小管间质性肾病 (ADTKD)、GUR (n = 137) 或未接受基因检测 (n = 1,076) 的家庭。测序,我们访问了 Genomics England 100,000 Genomes Project 数据。还鉴定出另外四个具有ALG5致病性变异的家族。23 名受影响成员的临床表现一致,囊性肾未肿大,肝囊肿很少或没有;8 名受试者在 62 岁至 91 岁期间罹患终末期肾病。我们证明ALG5单倍体不足足以改变肾上皮细胞中 N-聚糖链的合成。我们还表明 ALG5 是 PC1 成熟以及膜和纤毛定位所必需的,并且 ALG5 的杂合缺失会影响 PC1 成熟。总体而言,我们的结果表明,ALG5的单等位基因变异会导致 ADPKD 谱系疾病,其特征是多个小肾囊肿、进行性间质纤维化和肾功能下降。

























 京公网安备 11010802027423号
京公网安备 11010802027423号