当前位置:
X-MOL 学术
›
Acc. Mater. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A Convergent Understanding of Charged Defects
Accounts of Materials Research ( IF 14.6 ) Pub Date : 2022-06-21 , DOI: 10.1021/accountsmr.2c00044 Shashwat Anand 1, 2 , Michael Y. Toriyama 1 , Chris Wolverton 1 , Sossina M. Haile 1 , G. Jeffrey Snyder 1
Accounts of Materials Research ( IF 14.6 ) Pub Date : 2022-06-21 , DOI: 10.1021/accountsmr.2c00044 Shashwat Anand 1, 2 , Michael Y. Toriyama 1 , Chris Wolverton 1 , Sossina M. Haile 1 , G. Jeffrey Snyder 1
Affiliation
|
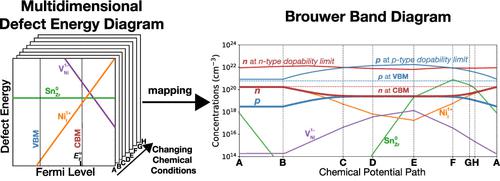
Historically, defects in semiconductors and ionic conductors have been studied using very different approaches. In the solid-state ionics community, nonstoichiometry and defect thermochemistry are often probed directly through experiments. The dependency of defect concentrations on chemical conditions (typically oxygen pressure) are modeled using a physical chemistry framework and compactly represented by the well-known Brouwer diagrams. In contrast, defects in electronic materials are now studied primarily with computational approaches─often density functional theory (DFT)─based on semiconductor physics in which the energy of defect formation also has an explicit dependence on the Fermi level, making the defect energy diagram multidimensional. As charged defects begin to attract the attention of experts from both schools of thought for applications in thermoelectrics, solar cells, batteries, fuel cells, and other electrochemical devices, a consistent understanding of charged defects addressing the apparent gaps in the two approaches is necessary.
中文翻译:

带电缺陷的趋同理解
从历史上看,半导体和离子导体中的缺陷已经使用非常不同的方法进行了研究。在固态离子学界,非化学计量和缺陷热化学通常直接通过实验进行探索。缺陷浓度对化学条件(通常是氧气压力)的依赖性使用物理化学框架建模,并由著名的 Brouwer 图紧凑表示。相比之下,电子材料中的缺陷现在主要通过计算方法进行研究——通常是密度泛函理论 (DFT)——基于半导体物理学,其中缺陷形成的能量也明显依赖于费米能级,从而使缺陷能量图具有多维性.
更新日期:2022-06-21
中文翻译:
带电缺陷的趋同理解
从历史上看,半导体和离子导体中的缺陷已经使用非常不同的方法进行了研究。在固态离子学界,非化学计量和缺陷热化学通常直接通过实验进行探索。缺陷浓度对化学条件(通常是氧气压力)的依赖性使用物理化学框架建模,并由著名的 Brouwer 图紧凑表示。相比之下,电子材料中的缺陷现在主要通过计算方法进行研究——通常是密度泛函理论 (DFT)——基于半导体物理学,其中缺陷形成的能量也明显依赖于费米能级,从而使缺陷能量图具有多维性.


























 京公网安备 11010802027423号
京公网安备 11010802027423号