当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Identifying a Feasible Transition Pathway between Two Conformational States for a Protein
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-06-20 , DOI: 10.1021/acs.jctc.2c00390 Yao Li 1, 2 , Haipeng Gong 1, 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-06-20 , DOI: 10.1021/acs.jctc.2c00390 Yao Li 1, 2 , Haipeng Gong 1, 2
Affiliation
|
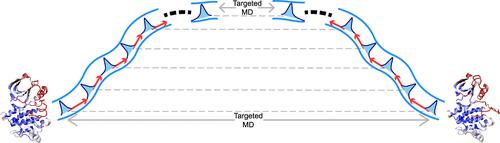
Proteins usually need to transit between different conformational states to fulfill their biological functions. In the mechanistic study of such transition processes by molecular dynamics simulations, identification of the minimum free energy path (MFEP) can substantially reduce the sampling space, thus enabling rigorous thermodynamic evaluation of the process. Conventionally, the MFEP is derived by iterative local optimization from an initial path, which is typically generated by simple brute force techniques like the targeted molecular dynamics (tMD). Therefore, the quality of the initial path determines the successfulness of MFEP estimation. In this work, we propose a method to improve derivation of the initial path. Through iterative relaxation-biasing simulations in a bidirectional manner, this method can construct a feasible transition pathway connecting two known states for a protein. Evaluation on small, fast-folding proteins against long equilibrium trajectories supports the good sampling efficiency of our method. When applied to larger proteins including the catalytic domain of human c-Src kinase as well as the converter domain of myosin VI, the paths generated by our method deviate significantly from those computed with the generic tMD approach. More importantly, free energy profiles and intermediate states obtained from our paths exhibit remarkable improvements over those from tMD paths with respect to both physical rationality and consistency with a priori knowledge.
中文翻译:

确定蛋白质两种构象状态之间的可行过渡途径
蛋白质通常需要在不同的构象状态之间转换才能实现其生物学功能。在通过分子动力学模拟对这种转变过程的机理研究中,确定最小自由能路径(MFEP)可以大大减少采样空间,从而能够对过程进行严格的热力学评估。传统上,MFEP 是从初始路径通过迭代局部优化导出的,该初始路径通常由简单的蛮力技术生成,如目标分子动力学 (tMD)。因此,初始路径的质量决定了MFEP估计的成功与否。在这项工作中,我们提出了一种改进初始路径推导的方法。通过双向的迭代松弛偏置模拟,该方法可以构建一条连接蛋白质两个已知状态的可行过渡途径。针对长平衡轨迹对小型快速折叠蛋白质的评估支持我们方法的良好采样效率。当应用于更大的蛋白质,包括人类 c-Src 激酶的催化结构域以及肌球蛋白 VI 的转换器结构域时,我们的方法生成的路径明显偏离使用通用 tMD 方法计算的路径。更重要的是,从我们的路径获得的自由能分布和中间状态在物理合理性和与先验知识的一致性方面表现出比从 tMD 路径获得的显着改进。针对长平衡轨迹的快速折叠蛋白质支持我们方法的良好采样效率。当应用于更大的蛋白质,包括人类 c-Src 激酶的催化域以及肌球蛋白 VI 的转换器域时,我们的方法生成的路径与使用通用 tMD 方法计算的路径显着偏离。更重要的是,从我们的路径获得的自由能分布和中间状态在物理合理性和与先验知识的一致性方面表现出比从 tMD 路径获得的显着改进。针对长平衡轨迹的快速折叠蛋白质支持我们方法的良好采样效率。当应用于更大的蛋白质,包括人类 c-Src 激酶的催化结构域以及肌球蛋白 VI 的转换器结构域时,我们的方法生成的路径明显偏离使用通用 tMD 方法计算的路径。更重要的是,从我们的路径获得的自由能分布和中间状态在物理合理性和与先验知识的一致性方面表现出比从 tMD 路径获得的显着改进。
更新日期:2022-06-20
中文翻译:
确定蛋白质两种构象状态之间的可行过渡途径
蛋白质通常需要在不同的构象状态之间转换才能实现其生物学功能。在通过分子动力学模拟对这种转变过程的机理研究中,确定最小自由能路径(MFEP)可以大大减少采样空间,从而能够对过程进行严格的热力学评估。传统上,MFEP 是从初始路径通过迭代局部优化导出的,该初始路径通常由简单的蛮力技术生成,如目标分子动力学 (tMD)。因此,初始路径的质量决定了MFEP估计的成功与否。在这项工作中,我们提出了一种改进初始路径推导的方法。通过双向的迭代松弛偏置模拟,该方法可以构建一条连接蛋白质两个已知状态的可行过渡途径。针对长平衡轨迹对小型快速折叠蛋白质的评估支持我们方法的良好采样效率。当应用于更大的蛋白质,包括人类 c-Src 激酶的催化结构域以及肌球蛋白 VI 的转换器结构域时,我们的方法生成的路径明显偏离使用通用 tMD 方法计算的路径。更重要的是,从我们的路径获得的自由能分布和中间状态在物理合理性和与先验知识的一致性方面表现出比从 tMD 路径获得的显着改进。针对长平衡轨迹的快速折叠蛋白质支持我们方法的良好采样效率。当应用于更大的蛋白质,包括人类 c-Src 激酶的催化域以及肌球蛋白 VI 的转换器域时,我们的方法生成的路径与使用通用 tMD 方法计算的路径显着偏离。更重要的是,从我们的路径获得的自由能分布和中间状态在物理合理性和与先验知识的一致性方面表现出比从 tMD 路径获得的显着改进。针对长平衡轨迹的快速折叠蛋白质支持我们方法的良好采样效率。当应用于更大的蛋白质,包括人类 c-Src 激酶的催化结构域以及肌球蛋白 VI 的转换器结构域时,我们的方法生成的路径明显偏离使用通用 tMD 方法计算的路径。更重要的是,从我们的路径获得的自由能分布和中间状态在物理合理性和与先验知识的一致性方面表现出比从 tMD 路径获得的显着改进。

























 京公网安备 11010802027423号
京公网安备 11010802027423号