当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Force-Based Method to Determine the Potential Dependence in Electrochemical Barriers
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2022-06-17 , DOI: 10.1021/acs.jpclett.2c01367 Sudarshan Vijay 1 , Georg Kastlunger 1 , Joseph A Gauthier 2, 3 , Anjli Patel 4 , Karen Chan 1
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2022-06-17 , DOI: 10.1021/acs.jpclett.2c01367 Sudarshan Vijay 1 , Georg Kastlunger 1 , Joseph A Gauthier 2, 3 , Anjli Patel 4 , Karen Chan 1
Affiliation
|
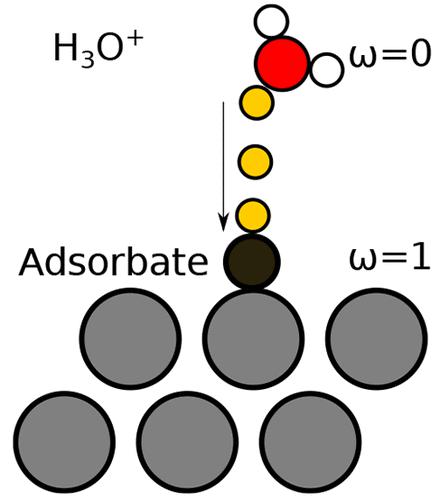
Determining ab initio potential-dependent energetics is critical to the investigation of mechanisms for electrochemical reactions. While methodology for evaluating reaction thermodynamics is established, simulation techniques for the corresponding kinetics is still a major challenge owing to a lack of potential control, finite cell size effects, or computational expense. In this work, we develop a model that allows for computing electrochemical activation energies from just a handful of density functional theory (DFT) calculations. The sole input into the model are the atom-centered forces obtained from DFT calculations performed on a homogeneous grid composed of varying field strengths. We show that the activation energies as a function of the potential obtained from our model are consistent for different supercell sizes and proton concentrations for a range of electrochemical reactions.
中文翻译:

确定电化学势垒中电位依赖性的基于力的方法
确定从头算电位依赖性能量学对于研究电化学反应机制至关重要。虽然建立了评估反应热力学的方法,但由于缺乏潜在的控制、有限的单元尺寸效应或计算费用,相应动力学的模拟技术仍然是一项重大挑战。在这项工作中,我们开发了一个模型,该模型允许仅通过少量密度泛函理论 (DFT) 计算来计算电化学活化能。模型的唯一输入是从由不同场强组成的均匀网格上执行的 DFT 计算获得的原子中心力。
更新日期:2022-06-17
中文翻译:
确定电化学势垒中电位依赖性的基于力的方法
确定从头算电位依赖性能量学对于研究电化学反应机制至关重要。虽然建立了评估反应热力学的方法,但由于缺乏潜在的控制、有限的单元尺寸效应或计算费用,相应动力学的模拟技术仍然是一项重大挑战。在这项工作中,我们开发了一个模型,该模型允许仅通过少量密度泛函理论 (DFT) 计算来计算电化学活化能。模型的唯一输入是从由不同场强组成的均匀网格上执行的 DFT 计算获得的原子中心力。

























 京公网安备 11010802027423号
京公网安备 11010802027423号