当前位置:
X-MOL 学术
›
J. Mater. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Data mining for predicting gas diffusivity in zeolitic-imidazolate frameworks (ZIFs)
Journal of Materials Chemistry A ( IF 11.9 ) Pub Date : 2022-06-16 , DOI: 10.1039/d2ta02624d Panagiotis Krokidas 1 , Stelios Karozis 2 , Salvador Moncho 3 , George Giannakopoulos 4, 5 , Edward N. Brothers 3 , Michael E. Kainourgiakis 2 , Ioannis G. Economou 1, 6 , Theodore A. Steriotis 1
Journal of Materials Chemistry A ( IF 11.9 ) Pub Date : 2022-06-16 , DOI: 10.1039/d2ta02624d Panagiotis Krokidas 1 , Stelios Karozis 2 , Salvador Moncho 3 , George Giannakopoulos 4, 5 , Edward N. Brothers 3 , Michael E. Kainourgiakis 2 , Ioannis G. Economou 1, 6 , Theodore A. Steriotis 1
Affiliation
|
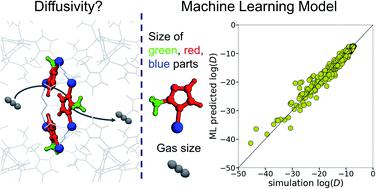
Molecular sieving is based on mobility differences of species under extreme confinement, i.e. within pores of molecular dimensions. The pore properties of a material determine its separation efficiency, while pore network engineering provides a way to optimize the sieving performance. Unlike rigid and structurally limited carbon and zeolite molecular sieves, metal organic frameworks (MOFs) offer flexible networks with unlimited pore tailoring possibilities, by using different linkers, functional groups and metals/clusters. Nevertheless, knowledge-based pore optimization towards highly selective materials is hampered by the complex relationship between structural modifications and molecular diffusivity. Machine learning (ML) approaches can elucidate this correlation, but pertinent research in MOFs has so far focused solely on sorption properties. Herein, we report the first ML-assisted work towards understanding how the replacement of basic MOF building units affects the pore structure and consequently the molecular diffusivity. The ML approach developed is general; the work is however focused on zeolitic-imidazolate frameworks (ZIFs) with SOD topology. Since there is no database of relevant ZIF variations, we constructed a new ensemble of 72 existing and new ZIFs through systematic sub-unit replacement, developed a force-field for each of these structures and performed molecular dynamics (MD) simulations on fully flexible systems to calculate framework properties and the diffusivity of different molecules (ranging from helium to n-butane). Based on this new database, a predictive multi-step ML model was developed and trained. The model can rapidly and efficiently estimate the diffusivity of molecules in any possible ZIF structure with SOD topology by using readily accessible input information.
中文翻译:

用于预测沸石-咪唑骨架 (ZIF) 中气体扩散率的数据挖掘
分子筛分是基于极端限制下物种的迁移率差异,即在分子尺寸的孔内。材料的孔隙特性决定了其分离效率,而孔隙网络工程提供了一种优化筛分性能的方法。与刚性和结构受限的碳和沸石分子筛不同,金属有机框架 (MOF) 通过使用不同的连接体、官能团和金属/簇,提供了具有无限孔定制可能性的灵活网络。然而,针对高选择性材料的基于知识的孔优化受到结构修饰和分子扩散率之间复杂关系的阻碍。机器学习 (ML) 方法可以阐明这种相关性,但迄今为止,MOF 的相关研究仅集中在吸附特性上。在此处,我们报告了第一个 ML 辅助工作,以了解基本 MOF 构建单元的替换如何影响孔结构,从而影响分子扩散率。开发的 ML 方法是通用的;然而,这项工作的重点是具有 SOD 拓扑结构的沸石-咪唑酯骨架 (ZIF)。由于没有相关 ZIF 变化的数据库,我们通过系统的子单元替换构建了一个包含 72 个现有和新 ZIF 的新集合,为这些结构中的每一个开发了力场,并在完全灵活的系统上进行了分子动力学 (MD) 模拟计算框架性质和不同分子的扩散率(从氦到 然而,这项工作的重点是具有 SOD 拓扑结构的沸石-咪唑酯骨架 (ZIF)。由于没有相关 ZIF 变化的数据库,我们通过系统的子单元替换构建了一个包含 72 个现有和新 ZIF 的新集合,为这些结构中的每一个开发了力场,并在完全灵活的系统上进行了分子动力学 (MD) 模拟计算框架性质和不同分子的扩散率(从氦到 然而,这项工作的重点是具有 SOD 拓扑结构的沸石-咪唑酯骨架 (ZIF)。由于没有相关 ZIF 变化的数据库,我们通过系统的子单元替换构建了一个包含 72 个现有和新 ZIF 的新集合,为这些结构中的每一个开发了力场,并在完全灵活的系统上进行了分子动力学 (MD) 模拟计算框架性质和不同分子的扩散率(从氦到正丁烷)。基于这个新数据库,开发和训练了一个预测多步 ML 模型。该模型可以通过使用易于访问的输入信息快速有效地估计具有 SOD 拓扑的任何可能 ZIF 结构中分子的扩散率。
更新日期:2022-06-16
中文翻译:
用于预测沸石-咪唑骨架 (ZIF) 中气体扩散率的数据挖掘
分子筛分是基于极端限制下物种的迁移率差异,即在分子尺寸的孔内。材料的孔隙特性决定了其分离效率,而孔隙网络工程提供了一种优化筛分性能的方法。与刚性和结构受限的碳和沸石分子筛不同,金属有机框架 (MOF) 通过使用不同的连接体、官能团和金属/簇,提供了具有无限孔定制可能性的灵活网络。然而,针对高选择性材料的基于知识的孔优化受到结构修饰和分子扩散率之间复杂关系的阻碍。机器学习 (ML) 方法可以阐明这种相关性,但迄今为止,MOF 的相关研究仅集中在吸附特性上。在此处,我们报告了第一个 ML 辅助工作,以了解基本 MOF 构建单元的替换如何影响孔结构,从而影响分子扩散率。开发的 ML 方法是通用的;然而,这项工作的重点是具有 SOD 拓扑结构的沸石-咪唑酯骨架 (ZIF)。由于没有相关 ZIF 变化的数据库,我们通过系统的子单元替换构建了一个包含 72 个现有和新 ZIF 的新集合,为这些结构中的每一个开发了力场,并在完全灵活的系统上进行了分子动力学 (MD) 模拟计算框架性质和不同分子的扩散率(从氦到 然而,这项工作的重点是具有 SOD 拓扑结构的沸石-咪唑酯骨架 (ZIF)。由于没有相关 ZIF 变化的数据库,我们通过系统的子单元替换构建了一个包含 72 个现有和新 ZIF 的新集合,为这些结构中的每一个开发了力场,并在完全灵活的系统上进行了分子动力学 (MD) 模拟计算框架性质和不同分子的扩散率(从氦到 然而,这项工作的重点是具有 SOD 拓扑结构的沸石-咪唑酯骨架 (ZIF)。由于没有相关 ZIF 变化的数据库,我们通过系统的子单元替换构建了一个包含 72 个现有和新 ZIF 的新集合,为这些结构中的每一个开发了力场,并在完全灵活的系统上进行了分子动力学 (MD) 模拟计算框架性质和不同分子的扩散率(从氦到正丁烷)。基于这个新数据库,开发和训练了一个预测多步 ML 模型。该模型可以通过使用易于访问的输入信息快速有效地估计具有 SOD 拓扑的任何可能 ZIF 结构中分子的扩散率。

























 京公网安备 11010802027423号
京公网安备 11010802027423号