当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Manipulating Coordination Structures of Mixed-Valence Copper Single Atoms on 1T-MoS2 for Efficient Hydrogen Evolution
ACS Catalysis ( IF 12.9 ) Pub Date : 2022-06-14 , DOI: 10.1021/acscatal.2c00759 Zhida Li 1, 2 , Xingxu Yan 3 , Dong He 4 , Wenhui Hu 5 , Sabrina Younan 1 , Zunjian Ke 1, 4 , Margaret Patrick 1 , Xiangheng Xiao 4 , Jier Huang 5 , Hongjun Wu 1, 2 , Xiaoqing Pan 3, 6 , Jing Gu 1
ACS Catalysis ( IF 12.9 ) Pub Date : 2022-06-14 , DOI: 10.1021/acscatal.2c00759 Zhida Li 1, 2 , Xingxu Yan 3 , Dong He 4 , Wenhui Hu 5 , Sabrina Younan 1 , Zunjian Ke 1, 4 , Margaret Patrick 1 , Xiangheng Xiao 4 , Jier Huang 5 , Hongjun Wu 1, 2 , Xiaoqing Pan 3, 6 , Jing Gu 1
Affiliation
|
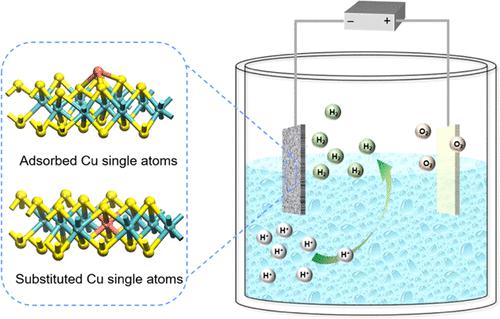
Directing single atoms (SAs) to occupy specific lattice sites in support materials and correlating the resulting changes in atomic coordination structures to catalytic activity are crucial for the rational design of high-performance single-atom catalysts (SACs). Herein, for the same copper (Cu) SAs, two coordination structures on 1T-phase molybdenum disulfide (MoS2) are identified. In the adsorption model, Cu SAs are coordinated onto the outermost sulfur (S) plane in a trigonal pyramidal geometry (Cuads@MoS2), while in the substitution model, Cu SAs are substituted with molybdenum (Mo) atoms (Cusub@MoS2), octahedrally coordinating with six S atoms in 1T-MoS2. Interestingly, in both coordination models, Cu(I) and Cu(II) SAs simultaneously exist. Cuads@MoS2 (173 mV) and Cusub@MoS2 (160 mV) demonstrate significantly reduced overpotential (at 10 mA cm–2) for the hydrogen evolution reaction (HER) compared to that of 1T-MoS2 (235 mV) under acidic condition (0.5 M H2SO4). Additionally, theoretical results reveal that different coordination structures would result in distinct active sites, where in Cuads@MoS2, the Cu SAs are the active sites, whereas in Cusub@MoS2, the S atoms most adjacent to the Cu SAs are the major active sites. Accurately determining the atomic coordination structures of SAs and uncovering their structure–property relationships are essential to guiding future geometry and structural research on SACs.
中文翻译:

操纵 1T-MoS2 上混合价铜单原子的配位结构以实现高效析氢
引导单原子(SAs)占据载体材料中的特定晶格位点,并将由此产生的原子配位结构变化与催化活性相关联,对于合理设计高性能单原子催化剂(SACs)至关重要。在此,对于相同的铜 (Cu) SA,确定了 1T 相二硫化钼 (MoS 2 ) 上的两个配位结构。在吸附模型中,Cu SAs 以三角锥几何形状(Cu ads @MoS 2)配位到最外层的硫 (S) 平面上,而在替代模型中,Cu SAs 被钼 (Mo) 原子取代(Cu sub @ MoS 2 ),八面体与 1T-MoS 2中的六个 S 原子配位. 有趣的是,在这两种配位模型中,Cu(I) 和 Cu(II) SAs 同时存在。与 1T-MoS 2 (235 mV ) 相比, Cu ads @MoS 2 (173 mV) 和 Cu sub @MoS 2 (160 mV) 表明析氢反应 (HER) 的过电势显着降低(在 10 mA cm –2下) ) 在酸性条件下 (0.5 MH 2 SO 4 )。此外,理论结果表明,不同的配位结构会导致不同的活性位点,其中在 Cu ads @MoS 2中,Cu SAs 是活性位点,而在 Cu sub @MoS 2中,最靠近Cu SAs的S原子是主要的活性位点。准确确定SAs的原子配位结构并揭示它们的结构-性质关系对于指导未来SACs的几何和结构研究至关重要。
更新日期:2022-06-14
中文翻译:
操纵 1T-MoS2 上混合价铜单原子的配位结构以实现高效析氢
引导单原子(SAs)占据载体材料中的特定晶格位点,并将由此产生的原子配位结构变化与催化活性相关联,对于合理设计高性能单原子催化剂(SACs)至关重要。在此,对于相同的铜 (Cu) SA,确定了 1T 相二硫化钼 (MoS 2 ) 上的两个配位结构。在吸附模型中,Cu SAs 以三角锥几何形状(Cu ads @MoS 2)配位到最外层的硫 (S) 平面上,而在替代模型中,Cu SAs 被钼 (Mo) 原子取代(Cu sub @ MoS 2 ),八面体与 1T-MoS 2中的六个 S 原子配位. 有趣的是,在这两种配位模型中,Cu(I) 和 Cu(II) SAs 同时存在。与 1T-MoS 2 (235 mV ) 相比, Cu ads @MoS 2 (173 mV) 和 Cu sub @MoS 2 (160 mV) 表明析氢反应 (HER) 的过电势显着降低(在 10 mA cm –2下) ) 在酸性条件下 (0.5 MH 2 SO 4 )。此外,理论结果表明,不同的配位结构会导致不同的活性位点,其中在 Cu ads @MoS 2中,Cu SAs 是活性位点,而在 Cu sub @MoS 2中,最靠近Cu SAs的S原子是主要的活性位点。准确确定SAs的原子配位结构并揭示它们的结构-性质关系对于指导未来SACs的几何和结构研究至关重要。


























 京公网安备 11010802027423号
京公网安备 11010802027423号