当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
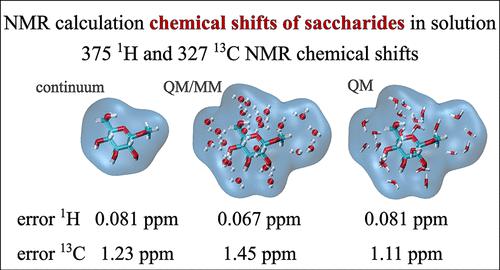
Efficiently Computing NMR 1H and 13C Chemical Shifts of Saccharides in Aqueous Environment
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-06-10 , DOI: 10.1021/acs.jctc.2c00127 Vladimír Palivec 1 , Radek Pohl 1 , Jakub Kaminský 1 , Hector Martinez-Seara 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-06-10 , DOI: 10.1021/acs.jctc.2c00127 Vladimír Palivec 1 , Radek Pohl 1 , Jakub Kaminský 1 , Hector Martinez-Seara 1
Affiliation
|
Determining the structure of saccharides in their native environment is crucial to understanding their function and more accurately targeting their utilization. Nuclear magnetic resonance observables such as the nuclear Overhauser effect or spin–spin coupling constants are routinely utilized to study saccharides in their native water environment. However, while highly sensitive to the local environment, chemical shifts are mostly overlooked, despite being commonly measured for compounds identification. Although chemical shifts carry considerable structural information, their direct association with structure is notoriously difficult. This is mostly due to the similarity in the chemical nature of most saccharides causing similar physicochemical environments close to sugar C and H atoms, resulting in comparable chemical shifts. The rise of computational power allows one to compute reliable chemical shifts and use them to determine atomistic details of these sugars in solution. However, any prediction is severely limited by the computational protocol used and its accuracy. In this work, we studied a set of 31 saccharides on which we evaluated various computational protocols to calculate the total number of 375 1H and 327 13C chemical shifts of sugars in an aqueous environment. Our study proposes two cost-effective protocols for simulating 1H and 13C chemical shifts that we recommend for further use. These protocols can help with the interpretation of experimental spectra, but we also show that they are also capable of structure prediction independently. This is possible because of the low mean absolute deviations of calculated shifts from the experiment (0.06 ppm for 1H and 1.09 ppm for 13C). We explore different solvation methods, basis sets, and optimization schemes to reach such accuracy. A correct sampling of the conformation phase space of flexible sugar molecules is also key to obtaining accurately converged theoretical chemical shifts. The linear regression method was applied to convert the calculated isotropic nuclear magnetic shielding constants to simulated chemical shifts comparable with the experiment. The achieved level of accuracy can help in utilizing chemical shifts for elucidating the 3D atomistic structure of saccharides in aqueous solutions. All linear regression parameters obtained on our extensive set of sugars for all the tested protocols can be reutilized in future works.
中文翻译:

高效计算水环境中糖的 NMR 1H 和 13C 化学位移
确定糖类在其原生环境中的结构对于了解它们的功能和更准确地定位它们的利用至关重要。核磁共振观测值,例如核 Overhauser 效应或自旋-自旋耦合常数,通常用于研究天然水环境中的糖类。然而,虽然对当地环境高度敏感,但化学位移大多被忽视,尽管通常用于化合物鉴定。尽管化学位移携带了大量的结构信息,但它们与结构的直接关联却是出了名的困难。这主要是由于大多数糖类的化学性质相似,导致靠近糖 C 和 H 原子的物理化学环境相似,从而产生可比较的化学位移。计算能力的提高使人们能够计算出可靠的化学位移,并使用它们来确定溶液中这些糖的原子细节。然而,任何预测都受到所使用的计算协议及其准确性的严重限制。在这项工作中,我们研究了一组 31 种糖类,我们在这些糖类上评估了各种计算协议以计算 375水环境中糖的1 H 和 327 13 C 化学位移。我们的研究提出了两种具有成本效益的方案来模拟1 H 和13 C 化学位移,我们建议进一步使用这些方案。这些协议可以帮助解释实验光谱,但我们也表明它们也能够独立地进行结构预测。这是可能的,因为从实验计算的偏移的平均绝对偏差很小(1 H 为 0.06 ppm, 13为 1.09 ppmC)。我们探索不同的求解方法、基组和优化方案以达到这种精度。对柔性糖分子的构象相空间进行正确采样也是获得准确收敛的理论化学位移的关键。应用线性回归方法将计算的各向同性核磁屏蔽常数转换为与实验相当的模拟化学位移。所达到的准确度水平有助于利用化学位移来阐明水溶液中糖类的 3D 原子结构。对于所有测试的协议,在我们广泛的糖组上获得的所有线性回归参数都可以在未来的工作中重新使用。
更新日期:2022-06-10
中文翻译:
高效计算水环境中糖的 NMR 1H 和 13C 化学位移
确定糖类在其原生环境中的结构对于了解它们的功能和更准确地定位它们的利用至关重要。核磁共振观测值,例如核 Overhauser 效应或自旋-自旋耦合常数,通常用于研究天然水环境中的糖类。然而,虽然对当地环境高度敏感,但化学位移大多被忽视,尽管通常用于化合物鉴定。尽管化学位移携带了大量的结构信息,但它们与结构的直接关联却是出了名的困难。这主要是由于大多数糖类的化学性质相似,导致靠近糖 C 和 H 原子的物理化学环境相似,从而产生可比较的化学位移。计算能力的提高使人们能够计算出可靠的化学位移,并使用它们来确定溶液中这些糖的原子细节。然而,任何预测都受到所使用的计算协议及其准确性的严重限制。在这项工作中,我们研究了一组 31 种糖类,我们在这些糖类上评估了各种计算协议以计算 375水环境中糖的1 H 和 327 13 C 化学位移。我们的研究提出了两种具有成本效益的方案来模拟1 H 和13 C 化学位移,我们建议进一步使用这些方案。这些协议可以帮助解释实验光谱,但我们也表明它们也能够独立地进行结构预测。这是可能的,因为从实验计算的偏移的平均绝对偏差很小(1 H 为 0.06 ppm, 13为 1.09 ppmC)。我们探索不同的求解方法、基组和优化方案以达到这种精度。对柔性糖分子的构象相空间进行正确采样也是获得准确收敛的理论化学位移的关键。应用线性回归方法将计算的各向同性核磁屏蔽常数转换为与实验相当的模拟化学位移。所达到的准确度水平有助于利用化学位移来阐明水溶液中糖类的 3D 原子结构。对于所有测试的协议,在我们广泛的糖组上获得的所有线性回归参数都可以在未来的工作中重新使用。
























 京公网安备 11010802027423号
京公网安备 11010802027423号