European Journal of Medicinal Chemistry ( IF 6.7 ) Pub Date : 2022-06-07 , DOI: 10.1016/j.ejmech.2022.114492 Shi Ding 1 , Ziye Gao 2 , Ziqiang Hu 2 , Rui Qi 2 , Xiangshan Zheng 2 , Xiaoyong Dong 2 , Mingjuan Zhang 2 , Jiwei Shen 1 , Tian Long 2 , Yan Zhu 2 , Lu Tian 2 , Wenshan Song 2 , Ruoqing Liu 2 , Ying Li 2 , Jiahuan Sun 2 , Wenwen Duan 3 , Ju Liu 1 , Ye Chen 1
|
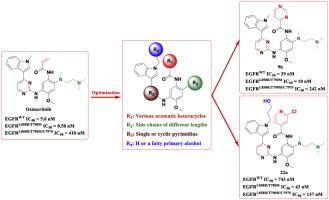
A series of osimertinib derivatives without acrylamide groups were synthesized and their inhibitory rates against L858R/T790M/C797S mutated EGFR kinase and antiproliferation activities against non-small cell lung cancer cell lines (A549, H1975) were evaluated. The preferred compounds were selected and their in vitro inhibitory activities against various EGFR kinases (wild-type, L858R/T790M, L858R/T790M/C797S) and c-Met kinase were tested. Compound 9h showed remarkable inhibitory activity against the wild type (IC50 = 29 nM), L858R/T790M mutant type (IC50 = 10 nM) and L858R/T790M/C797S mutant type (IC50 = 242 nM) as reversible EGFR kinase inhibitor, which was selected to further perform the AO/EB staining assays, cell cycle distribution assays and wound-healing assays on A549 and/or H1975 cell lines. The results showed dose-dependent activities of the induction of cell apoptosis, G1/G0-phase arrestation and inhibition of migration. Compound 22a showed remarkable inhibitory activity against the L858R/T790M/C797S mutant EGFR kinase (IC50 = 137 nM), which was nearly three times compared to osimertinib (IC50 = 410 nM). It's worth noting that 22a exhibited excellent kinase selectivity against the L858R/T790M/C797S mutant EGFR kinase rather than the wild-type, which reached 5.4 times and far more than the 0.012 times of osimertinib. Additionally, molecular docking analyses were performed to explain the action modes between the compounds and the corresponding EGFR kinases. In conclusion, compounds 9h and 22a have been demonstrated as promising candidates and worth further study.
中文翻译:
作为可逆 EGFR 激酶抑制剂的新型奥希替尼衍生物的设计、合成和生物学评价
合成了一系列不含丙烯酰胺基团的奥希替尼衍生物,评估了它们对L858R/T790M/C797S突变EGFR激酶的抑制率和对非小细胞肺癌细胞系(A549、H1975)的抗增殖活性。选择了优选的化合物,并测试了它们对各种 EGFR 激酶(野生型、L858R/T790M、L858R/T790M/C797S)和 c-Met 激酶的体外抑制活性。化合物9h对野生型 (IC 50 = 29 nM)、L858R/T790M 突变型 (IC 50 = 10 nM) 和 L858R/T790M/C797S 突变型 (IC 50 = 242 nM) 作为可逆的 EGFR 激酶抑制剂,被选择用于进一步对 A549 和/或 H1975 细胞系进行 AO/EB 染色测定、细胞周期分布测定和伤口愈合测定。结果显示诱导细胞凋亡、G1/G0期停滞和抑制迁移的剂量依赖性活性。化合物22a显示出对 L858R/T790M/C797S 突变 EGFR 激酶的显着抑制活性 (IC 50 = 137 nM),与奥希替尼 (IC 50 = 410 nM) 相比几乎是三倍。值得注意的是,22a对 L858R/T790M/C797S 突变型 EGFR 激酶而非野生型表现出优异的激酶选择性,达到 5.4 倍,远超过奥希替尼的 0.012 倍。此外,进行分子对接分析以解释化合物与相应 EGFR 激酶之间的作用模式。总之,化合物9h和22a已被证明是有前途的候选者,值得进一步研究。

























 京公网安备 11010802027423号
京公网安备 11010802027423号