当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Explicitly Correlated Electronic Structure Calculations with Transcorrelated Matrix Product Operators
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-06-06 , DOI: 10.1021/acs.jctc.2c00167 Alberto Baiardi 1 , Michał Lesiuk 1, 2 , Markus Reiher 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-06-06 , DOI: 10.1021/acs.jctc.2c00167 Alberto Baiardi 1 , Michał Lesiuk 1, 2 , Markus Reiher 1
Affiliation
|
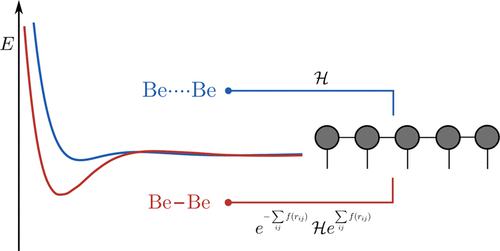
In this work, we present the first implementation of the transcorrelated electronic Hamiltonian in an optimization procedure for matrix product states by the density matrix renormalization group (DMRG) algorithm. In the transcorrelation ansatz, the electronic Hamiltonian is similarity-transformed with a Jastrow factor to describe the cusp in the wave function at electron–electron coalescence. As a result, the wave function is easier to approximate accurately with the conventional expansion in terms of one-particle basis functions and Slater determinants. The transcorrelated Hamiltonian in first quantization comprises up to three-body interactions, which we deal with in the standard way by applying robust density fitting to two- and three-body integrals entering the second-quantized representation of this Hamiltonian. The lack of hermiticity of the transcorrelated Hamiltonian is taken care of along the lines of the first work on transcorrelated DMRG [ J. Chem. Phys. 2020, 153, 164115] by encoding it as a matrix product operator and optimizing the corresponding ground state wave function with imaginary-time time-dependent DMRG. We demonstrate our quantum chemical transcorrelated DMRG approach at the example of several atoms and first-row diatomic molecules. We show that transcorrelation improves the convergence rate to the complete basis set limit in comparison to conventional DMRG. Moreover, we study extensions of our approach that aim at reducing the cost of handling the matrix product operator representation of the transcorrelated Hamiltonian.
中文翻译:

具有转相关矩阵乘积算子的显式相关电子结构计算
在这项工作中,我们通过密度矩阵重整化组 (DMRG) 算法在矩阵乘积状态的优化过程中首次实现了跨相关电子哈密顿量。在互相关分析中,电子哈密顿量用 Jastrow 因子进行相似变换,以描述电子 - 电子聚结时波函数的尖点。因此,在单粒子基函数和 Slater 行列式方面,波函数更容易用常规展开式精确逼近。第一次量化中的转相关哈密顿量最多包含三体相互作用,我们通过对进入该哈密顿量的第二次量化表示的二体和三体积分应用稳健的密度拟合来以标准方式处理这些相互作用。J.化学。物理。 2020 , 153 , 164115] 通过将其编码为矩阵乘积算子并使用虚时随时间的 DMRG 优化相应的基态波函数。我们在几个原子和第一行双原子分子的例子中展示了我们的量子化学反相关 DMRG 方法。我们表明,与传统的 DMRG 相比,互相关将收敛速度提高到了完整的基组限制。此外,我们研究了旨在降低处理转相关哈密顿量的矩阵乘积算子表示的成本的方法的扩展。
更新日期:2022-06-06
中文翻译:
具有转相关矩阵乘积算子的显式相关电子结构计算
在这项工作中,我们通过密度矩阵重整化组 (DMRG) 算法在矩阵乘积状态的优化过程中首次实现了跨相关电子哈密顿量。在互相关分析中,电子哈密顿量用 Jastrow 因子进行相似变换,以描述电子 - 电子聚结时波函数的尖点。因此,在单粒子基函数和 Slater 行列式方面,波函数更容易用常规展开式精确逼近。第一次量化中的转相关哈密顿量最多包含三体相互作用,我们通过对进入该哈密顿量的第二次量化表示的二体和三体积分应用稳健的密度拟合来以标准方式处理这些相互作用。J.化学。物理。 2020 , 153 , 164115] 通过将其编码为矩阵乘积算子并使用虚时随时间的 DMRG 优化相应的基态波函数。我们在几个原子和第一行双原子分子的例子中展示了我们的量子化学反相关 DMRG 方法。我们表明,与传统的 DMRG 相比,互相关将收敛速度提高到了完整的基组限制。此外,我们研究了旨在降低处理转相关哈密顿量的矩阵乘积算子表示的成本的方法的扩展。


























 京公网安备 11010802027423号
京公网安备 11010802027423号