当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Insights from density functional theory calculations into the effects of the adsorption and dissociation of water on the surface properties of zinc diphosphide (ZnP2) nanocrystals
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2021-11-11 , DOI: 10.1039/d1cp02784k Barbara Farkaš 1 , Aleksandar Živković 1, 2 , Veikko Uahengo 3 , Nelson Y Dzade 1, 4 , Nora H de Leeuw 1, 2, 5
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2021-11-11 , DOI: 10.1039/d1cp02784k Barbara Farkaš 1 , Aleksandar Živković 1, 2 , Veikko Uahengo 3 , Nelson Y Dzade 1, 4 , Nora H de Leeuw 1, 2, 5
Affiliation
|
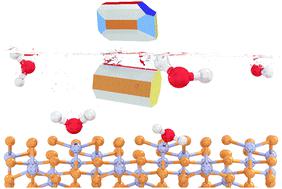
Zinc phosphides (ZnP2 and Zn3P2) are emerging absorber materials for photovoltaic applications owing to their abundancy and non-toxic nature. Herein, we provide a comprehensive characterisation of the surface structure, composition, stabilities, morphology, and electronic properties of both bare and hydrated/hydroxylated low-Miller index surfaces of β-ZnP2 by means of density functional theory (DFT) calculations. Mechanistic insights into the fundamental aspects of water adsorption and dissociation, including the adsorption geometries, energetics, and structural parameters along the reaction path are systematically characterised. The stabilities of the surfaces under dry and wet conditions are discussed in detail and the predicted phase diagrams for the water adsorption are presented. Using calculated surface energies, we have derived the equilibrium morphology of the β-ZnP2 nanocrystals under vacuum and upon hydration or hydroxylation. Atomic-level insights into the origin of the incipient oxidation of β-ZnP2 surfaces are provided through analysis of Bader charges, which reveal that the Zn sites to which H2O and OH species are bound undergo oxidation due to the transfer of charge to the adsorbed species. Adsorption-induced changes to the electronic properties before and after hydration/hydroxylation were characterised by the work function and partial density of states. The results highlight the need for protection of β-ZnP2 nanocrystals against possible oxidation in the presence of water through post-synthesis organic functionalisation.
中文翻译:

从密度泛函理论计算洞察水的吸附和解离对二磷化锌 (ZnP2) 纳米晶体表面性质的影响
磷化锌(ZnP 2和Zn 3 P 2)由于其丰富且无毒的性质而成为用于光伏应用的新兴吸收材料。在此,我们提供了 β-ZnP 2 的裸露和水合/羟基化低米勒指数表面的表面结构、组成、稳定性、形态和电子特性的综合表征通过密度泛函理论 (DFT) 计算。系统地表征了对水吸附和解离基本方面的机械见解,包括沿反应路径的吸附几何学、能量学和结构参数。详细讨论了干湿条件下表面的稳定性,并给出了预测的水吸附相图。使用计算的表面能,我们推导出了 β-ZnP 2纳米晶体在真空下和水合或羟基化时的平衡形态。通过对 Bader 电荷的分析,提供了对 β-ZnP 2表面初始氧化起源的原子级见解,这表明 H 2由于电荷转移到吸附物质上,O 和 OH 物质被结合而发生氧化。水合/羟基化前后吸附引起的电子性质变化的特征在于功函数和部分态密度。结果强调需要通过合成后的有机功能化来保护 β-ZnP 2纳米晶体在水存在下免受可能的氧化。
更新日期:2021-11-22
中文翻译:
从密度泛函理论计算洞察水的吸附和解离对二磷化锌 (ZnP2) 纳米晶体表面性质的影响
磷化锌(ZnP 2和Zn 3 P 2)由于其丰富且无毒的性质而成为用于光伏应用的新兴吸收材料。在此,我们提供了 β-ZnP 2 的裸露和水合/羟基化低米勒指数表面的表面结构、组成、稳定性、形态和电子特性的综合表征通过密度泛函理论 (DFT) 计算。系统地表征了对水吸附和解离基本方面的机械见解,包括沿反应路径的吸附几何学、能量学和结构参数。详细讨论了干湿条件下表面的稳定性,并给出了预测的水吸附相图。使用计算的表面能,我们推导出了 β-ZnP 2纳米晶体在真空下和水合或羟基化时的平衡形态。通过对 Bader 电荷的分析,提供了对 β-ZnP 2表面初始氧化起源的原子级见解,这表明 H 2由于电荷转移到吸附物质上,O 和 OH 物质被结合而发生氧化。水合/羟基化前后吸附引起的电子性质变化的特征在于功函数和部分态密度。结果强调需要通过合成后的有机功能化来保护 β-ZnP 2纳米晶体在水存在下免受可能的氧化。


























 京公网安备 11010802027423号
京公网安备 11010802027423号