Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2021-11-17 , DOI: 10.1016/j.csbj.2021.11.015 Mariana H Moreira 1 , Fabio C L Almeida 1 , Tatiana Domitrovic 2 , Fernando L Palhano 1
|
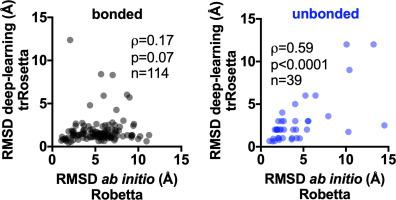
Defensins are small proteins, usually ranging from 3 to 6 kDa, amphipathic, disulfide-rich, and with a small or even absent hydrophobic core. Since a hydrophobic core is generally found in globular proteins that fold in an aqueous solvent, the peculiar fold of defensins can challenge tertiary protein structure predictors. We performed a Protein Data Bank survey of small proteins (3-6 kDa) to understand the similarities of defensins with other small disulfide-rich proteins. We found no differences when we compared defensins with non-defensins regarding the proportion of apolar, polar and charged residues and their exposure to the solvent. Then we divided all small proteins (3-6 kDa) in the Protein Data Bank into two groups, one group with at least one disulfide bond (bonded, defensins included) and another group without any disulfide bond (unbonded). The group of bonded proteins contained apolar residues more exposed to the solvent than the unbonded group. The ab initio algorithm for tertiary protein structure prediction Robetta was more accurate at predicting unbonded than bonded proteins. On the other hand, the trRosetta algorithm, which uses artificial intelligence, improved the prediction of most bonded proteins, while for the unbonded group no improvement was obtained. Our work highlights one more layer of complexity for the prediction of protein tertiary structure: small disulfide-rich proteins' ability to fold even with a poorly hydrophobic core.
中文翻译:
沉积在 PDB 中的所有已解决小蛋白的系统结构比较。二硫键在蛋白质折叠中的作用
防御素是小的蛋白质,通常在 3 到 6 kDa 的范围内,两亲的,富含二硫键,并且具有小的甚至没有疏水核心。由于疏水核心通常存在于在水性溶剂中折叠的球状蛋白质中,因此防御素的特殊折叠可能会挑战三级蛋白质结构预测因子。我们对小蛋白质 (3-6 kDa) 进行了蛋白质数据库调查,以了解防御素与其他富含二硫化物的小蛋白质的相似性。当我们比较防御素和非防御素关于非极性、极性和带电残基的比例及其对溶剂的暴露时,我们发现没有差异。然后我们将蛋白质数据库中的所有小蛋白质(3-6 kDa)分成两组,一组至少有一个二硫键(键合,包括防御素)和另一个没有任何二硫键的基团(未键合的)。与未键合基团相比,键合蛋白质组含有更多暴露于溶剂的非极性残基。这用于三级蛋白质结构预测的从头算算法 Robetta 在预测非键合蛋白质方面比键合蛋白质更准确。另一方面,使用人工智能的 trRosetta 算法改进了对大多数键合蛋白的预测,而对于未键合组则没有任何改进。我们的工作突出了预测蛋白质三级结构的另一层复杂性:即使是疏水性差的核心,富含二硫键的小蛋白质也能折叠。
























 京公网安备 11010802027423号
京公网安备 11010802027423号