当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Quasiclassical Direct Dynamics Trajectory Simulations of Organometallic Reactions
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2021-11-11 , DOI: 10.1021/acs.accounts.1c00575 Daniel H Ess 1
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2021-11-11 , DOI: 10.1021/acs.accounts.1c00575 Daniel H Ess 1
Affiliation
|
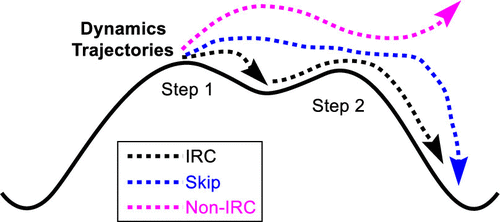
Homogeneous metal-mediated organometallic reactions represent a very large and diverse reaction class. Density functional theory calculations are now routinely carried out and reported for analyzing organometallic mechanisms and reaction pathways. While density functional theory calculations are extremely powerful to understand the energy and structure of organometallic reactions, there are several assumptions in their use and interpretation to define reaction mechanisms and to analyze reaction selectivity. Almost always it is assumed that potential energy structures calculated with density functional theory adequately describe mechanisms and selectivity within the framework of statistical theories, for example, transition state theory and RRKM theory. However, these static structures and corresponding energy landscapes do not provide atomic motion information during reactions that could reveal nonstatistical intermediates without complete intramolecular vibrational redistribution and nonintrinsic reaction coordinate (non-IRC) pathways. While nonstatistical intermediates and non-IRC reaction pathways are now relatively well established for organic reactions, these dynamic effects have heretofore been highly underexplored in organometallic reactions. Through a series of quasiclassical density functional theory direct dynamics trajectory studies, my group has recently demonstrated that dynamic effects occur in a variety of fundamental organometallic reactions, especially bond activation reactions. For example, in the C–H activation reaction between methane and [Cp*(PMe3)IrIII(CH3)]+, while the density functional theory energy landscape showed a two-step oxidative cleavage and reductive coupling mechanism, trajectories revealed a mixture of this two-step mechanism and a dynamic one-step mechanism that skipped the [Cp*(PMe3)IrV(H)(CH3)2]+ intermediate. This study also showed that despite a methane σ-complex being located on the density functional theory surface before oxidative cleavage and after reductive coupling, this intermediate is always skipped and should not be considered an intermediate during reactive trajectories. For non-IRC reaction pathways, quasiclassical direct dynamics trajectories showed that for the isomerization of [Tp(NO)(PMe3)W(η2-benzene)] to [Tp(NO)(PMe3)W(H)(Ph)], there are many dynamic reaction pathway connections due to a relatively flat energy landscape and π coordination is not necessary for C–H bond activation through oxidative cleavage. Trajectories also showed that dynamic effects are important in selectivity for ethylene C–H activation versus π coordination in reaction with Cp(PMe3)2Re, and trajectories provide a more quantitative model of selectivity than transition state theory. Quasiclassical trajectories examining Au-catalyzed monoallylic diol cyclizations showed dynamic coupling of several reaction steps that include alkoxylation π bond addition, proton shuttling, and water elimination reaction steps. Overall, these studies highlight the need to use direct dynamics trajectory simulations to consider atomic motion during reactions to understand organometallic reaction mechanisms and selectivity.
中文翻译:

有机金属反应的准经典直接动力学轨迹模拟
均相金属介导的有机金属反应代表了一个非常大且多样化的反应类别。现在常规进行密度泛函理论计算并报告用于分析有机金属机制和反应途径。虽然密度泛函理论计算对于理解有机金属反应的能量和结构非常有效,但在定义反应机制和分析反应选择性时,它们的使用和解释有几个假设。几乎总是假设用密度泛函理论计算的势能结构充分描述了统计理论框架内的机制和选择性,例如过渡态理论和 RRKM 理论。然而,这些静态结构和相应的能量图在反应过程中不提供原子运动信息,这些信息可以揭示没有完整分子内振动再分布和非固有反应坐标(非 IRC)路径的非统计中间体。虽然非统计中间体和非 IRC 反应途径现在已经相对完善地用于有机反应,但迄今为止,这些动态效应在有机金属反应中的探索还很不足。通过一系列准经典密度泛函理论直接动力学轨迹研究,我组最近证明了动力学效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe 虽然非统计中间体和非 IRC 反应途径现在已经相对完善地用于有机反应,但迄今为止,这些动态效应在有机金属反应中的探索还很不足。通过一系列准经典密度泛函理论直接动力学轨迹研究,我组最近证明了动力学效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe 虽然非统计中间体和非 IRC 反应途径现在已经相对完善地用于有机反应,但迄今为止,这些动态效应在有机金属反应中的探索还很不足。通过一系列准经典密度泛函理论直接动力学轨迹研究,我组最近证明了动力学效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe 我的小组最近证明,动态效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe 我的小组最近证明,动态效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe3 )Ir III (CH 3 )] +,虽然密度泛函理论能量图显示了两步氧化裂解和还原耦合机制,但轨迹显示了这种两步机制和动态一步机制的混合,跳过了[Cp*(PMe 3 )Ir V (H)(CH 3 ) 2 ] +中间的。该研究还表明,尽管甲烷 σ 配合物在氧化裂解之前和还原偶联之后位于密度泛函理论表面上,但该中间体总是被跳过,不应被视为反应轨迹中的中间体。对于非 IRC 反应途径,准经典直接动力学轨迹表明,对于 [Tp(NO)(PMe 3 )W(η 2 -苯)]异构化为[Tp(NO)(PMe 3)W(H)(Ph)],由于相对平坦的能量景观,存在许多动态反应途径连接,并且通过氧化裂解激活 C-H 键不需要 π 配位。轨迹还表明,动态效应在乙烯 C-H 活化与与 Cp(PMe 3 ) 2反应中的 π 配位的选择性方面很重要Re和轨迹提供了比过渡态理论更定量的选择性模型。检查金催化的单烯丙基二醇环化的准经典轨迹显示了几个反应步骤的动态耦合,包括烷氧基化 π 键加成、质子穿梭和水消除反应步骤。总体而言,这些研究强调需要使用直接动力学轨迹模拟来考虑反应过程中的原子运动,以了解有机金属反应机制和选择性。
更新日期:2021-12-07
中文翻译:
有机金属反应的准经典直接动力学轨迹模拟
均相金属介导的有机金属反应代表了一个非常大且多样化的反应类别。现在常规进行密度泛函理论计算并报告用于分析有机金属机制和反应途径。虽然密度泛函理论计算对于理解有机金属反应的能量和结构非常有效,但在定义反应机制和分析反应选择性时,它们的使用和解释有几个假设。几乎总是假设用密度泛函理论计算的势能结构充分描述了统计理论框架内的机制和选择性,例如过渡态理论和 RRKM 理论。然而,这些静态结构和相应的能量图在反应过程中不提供原子运动信息,这些信息可以揭示没有完整分子内振动再分布和非固有反应坐标(非 IRC)路径的非统计中间体。虽然非统计中间体和非 IRC 反应途径现在已经相对完善地用于有机反应,但迄今为止,这些动态效应在有机金属反应中的探索还很不足。通过一系列准经典密度泛函理论直接动力学轨迹研究,我组最近证明了动力学效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe 虽然非统计中间体和非 IRC 反应途径现在已经相对完善地用于有机反应,但迄今为止,这些动态效应在有机金属反应中的探索还很不足。通过一系列准经典密度泛函理论直接动力学轨迹研究,我组最近证明了动力学效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe 虽然非统计中间体和非 IRC 反应途径现在已经相对完善地用于有机反应,但迄今为止,这些动态效应在有机金属反应中的探索还很不足。通过一系列准经典密度泛函理论直接动力学轨迹研究,我组最近证明了动力学效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe 我的小组最近证明,动态效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe 我的小组最近证明,动态效应发生在各种基本的有机金属反应中,尤其是键活化反应。例如,在甲烷和 [Cp*(PMe3 )Ir III (CH 3 )] +,虽然密度泛函理论能量图显示了两步氧化裂解和还原耦合机制,但轨迹显示了这种两步机制和动态一步机制的混合,跳过了[Cp*(PMe 3 )Ir V (H)(CH 3 ) 2 ] +中间的。该研究还表明,尽管甲烷 σ 配合物在氧化裂解之前和还原偶联之后位于密度泛函理论表面上,但该中间体总是被跳过,不应被视为反应轨迹中的中间体。对于非 IRC 反应途径,准经典直接动力学轨迹表明,对于 [Tp(NO)(PMe 3 )W(η 2 -苯)]异构化为[Tp(NO)(PMe 3)W(H)(Ph)],由于相对平坦的能量景观,存在许多动态反应途径连接,并且通过氧化裂解激活 C-H 键不需要 π 配位。轨迹还表明,动态效应在乙烯 C-H 活化与与 Cp(PMe 3 ) 2反应中的 π 配位的选择性方面很重要Re和轨迹提供了比过渡态理论更定量的选择性模型。检查金催化的单烯丙基二醇环化的准经典轨迹显示了几个反应步骤的动态耦合,包括烷氧基化 π 键加成、质子穿梭和水消除反应步骤。总体而言,这些研究强调需要使用直接动力学轨迹模拟来考虑反应过程中的原子运动,以了解有机金属反应机制和选择性。
























 京公网安备 11010802027423号
京公网安备 11010802027423号