Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2021-11-05 , DOI: 10.1016/j.csbj.2021.11.005 Ajay Pal 1, 2 , James F Curtin 1 , Gemma K Kinsella 1
|
The G-protein coupled receptor, GPR120, has ubiquitous expression and multifaceted roles in modulating metabolic and anti-inflammatory processes. Recent implications of its role in cancer progression have presented GPR120 as an attractive oncogenic drug target. GPR120 gene knockdown in breast cancer studies revealed a role of GPR120-induced chemoresistance in epirubicin and cisplatin-induced DNA damage in tumour cells. Higher expression and activation levels of GPR120 is also reported to promote tumour angiogenesis and cell migration in colorectal cancer. Some agonists targeting GPR120 have been reported, such as TUG891 and Compound39, but to date development of small-molecule inhibitors of GPR120 is limited.
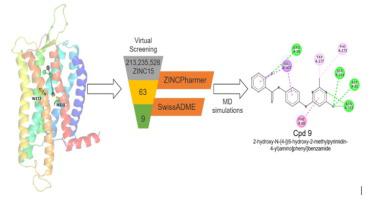
Herein, following homology modelling of the receptor a pharmacophore hypothesis was derived from 300ns all-atomic molecular dynamics (MD) simulations on apo, TUG891-bound and Compound39-bound GPR120S (short isoform) receptor models embedded in a water solvated lipid bilayer system. We performed comparative MD analysis on protein-ligand interactions between the two agonist and apo simulations on the stability of the “ionic lock” – a Class A GPCRs characteristic of receptor activation and inactivation. The detailed analysis predicted that ligand interactions with W277 and N313 are critical to conserve the “ionic-lock” conformation (R136 of Helix 3) and prevent GPR120S receptor activation. The results led to generation of a W277 and N313 focused pharmacophore hypothesis and the screening of the ZINC15 database using ZINCPharmer through the structure-based pharmacophore.
100ns all-atomic molecular dynamics (MD) simulations were performed on 9 small molecules identified and Cpd 9, (2-hydroxy-N-{4-[(6-hydroxy-2-methylpyrimidin-4-yl) amino] phenyl} benzamide) was predicted to be a small-molecule GPR120S antagonist. The conformational results from the collective all-atomic MD analysis provided structural information for further identification and optimisation of novel druggable inhibitors of GPR120S using this rational design approach, which could have future potential for anti-cancer drug development studies.
中文翻译:
基于药效团筛选和分子动力学模拟的新型 GPR120 拮抗剂的结构预测
G 蛋白偶联受体 GPR120 在调节代谢和抗炎过程中具有普遍的表达和多方面的作用。其在癌症进展中的作用的最新影响表明 GPR120 是一个有吸引力的致癌药物靶点。乳腺癌研究中的 GPR120 基因敲低揭示了 GPR120 诱导的化学抗性在表柔比星和顺铂诱导的肿瘤细胞 DNA 损伤中的作用。据报道,GPR120 的更高表达和激活水平可促进结直肠癌中的肿瘤血管生成和细胞迁移。已经报道了一些靶向 GPR120 的激动剂,例如 TUG891 和 Compound39,但迄今为止 GPR120 的小分子抑制剂的开发是有限的。
在本文中,根据受体的同源性建模,药效团假设源自对嵌入水溶剂化脂质双层系统中的载脂蛋白、TUG891 结合和化合物 39 结合 GPR120S(短同种型)受体模型的 300ns 全原子分子动力学 (MD) 模拟。我们对两种激动剂和载脂蛋白模拟之间的蛋白质-配体相互作用进行了比较 MD 分析,对“离子锁”的稳定性进行了模拟 - 受体激活和失活的 A 类 GPCR 特征。详细分析预测配体与 W277 和 N313 的相互作用对于保留“离子锁”构象(螺旋 3 的 R136)和防止 GPR120S 受体激活至关重要。
100ns 全原子分子动力学 (MD) 模拟对鉴定的 9 个小分子和 Cpd 9,(2-羟基-N-{4-[(6-羟基-2-甲基嘧啶-4-基)氨基]苯基}苯甲酰胺进行) 被预测为小分子 GPR120S 拮抗剂。集体全原子 MD 分析的构象结果为使用这种合理设计方法进一步鉴定和优化 GPR120S 的新型药物抑制剂提供了结构信息,这可能具有未来抗癌药物开发研究的潜力。

























 京公网安备 11010802027423号
京公网安备 11010802027423号