当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
An automatized workflow from molecular dynamic simulation to quantum chemical methods to identify elementary reactions and compute reaction constants
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2021-10-12 , DOI: 10.1002/jcc.26757 Gunnar Schmitz 1 , Özlem Yönder 2 , Bastian Schnieder 1 , Rochus Schmid 1 , Christof Hättig 2
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2021-10-12 , DOI: 10.1002/jcc.26757 Gunnar Schmitz 1 , Özlem Yönder 2 , Bastian Schnieder 1 , Rochus Schmid 1 , Christof Hättig 2
Affiliation
|
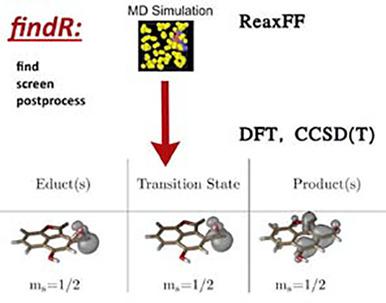
We present an automatized workflow which, starting from molecular dynamics simulations, identifies reaction events, filters them, and prepares them for accurate quantum chemical calculations using, for example, Density Functional Theory (DFT) or Coupled Cluster methods. The capabilities of the automatized workflow are demonstrated by the example of simulations for the combustion of some polycyclic aromatic hydrocarbons (PAHs). It is shown how key elementary reaction candidates are filtered out of a much larger set of redundant reactions and refined further. The molecular species in question are optimized using DFT and reaction energies, barrier heights, and reaction rates are calculated. The setup is general enough to include at this stage configurational sampling, which can be exploited in the future. Using the introduced machinery, we investigate how the observed reaction types depend on the gas atmosphere used in the molecular dynamics simulation. For the re-optimization on the DFT level, we show how the additional information needed to switch from reactive force-field to electronic structure calculations can be filled in and study how well ReaxFF and DFT agree with each other and shine light on the perspective of using more accurate semi-empirical methods in the MD simulation.
中文翻译:

从分子动力学模拟到量子化学方法的自动化工作流程,以识别基本反应和计算反应常数
我们提出了一个自动化的工作流程,从分子动力学模拟开始,识别反应事件,过滤它们,并使用例如密度泛函理论 (DFT) 或耦合聚类方法为准确的量子化学计算做好准备。一些多环芳烃 (PAH) 燃烧的模拟示例展示了自动化工作流程的功能。它显示了关键的基本反应候选者如何从更大的冗余反应集中过滤出来并进一步细化。所讨论的分子种类使用 DFT 进行优化,并计算反应能、势垒高度和反应速率。该设置足够通用,可以在此阶段包含配置采样,将来可以利用。使用引进的机器,我们研究观察到的反应类型如何取决于分子动力学模拟中使用的气体气氛。对于 DFT 级别的重新优化,我们展示了如何填写从反作用力场切换到电子结构计算所需的附加信息,并研究 ReaxFF 和 DFT 之间的一致性如何,并阐明了在 MD 模拟中使用更准确的半经验方法。
更新日期:2021-11-15
中文翻译:
从分子动力学模拟到量子化学方法的自动化工作流程,以识别基本反应和计算反应常数
我们提出了一个自动化的工作流程,从分子动力学模拟开始,识别反应事件,过滤它们,并使用例如密度泛函理论 (DFT) 或耦合聚类方法为准确的量子化学计算做好准备。一些多环芳烃 (PAH) 燃烧的模拟示例展示了自动化工作流程的功能。它显示了关键的基本反应候选者如何从更大的冗余反应集中过滤出来并进一步细化。所讨论的分子种类使用 DFT 进行优化,并计算反应能、势垒高度和反应速率。该设置足够通用,可以在此阶段包含配置采样,将来可以利用。使用引进的机器,我们研究观察到的反应类型如何取决于分子动力学模拟中使用的气体气氛。对于 DFT 级别的重新优化,我们展示了如何填写从反作用力场切换到电子结构计算所需的附加信息,并研究 ReaxFF 和 DFT 之间的一致性如何,并阐明了在 MD 模拟中使用更准确的半经验方法。


























 京公网安备 11010802027423号
京公网安备 11010802027423号