当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Quantum-Based Molecular Dynamics Simulations Using Tensor Cores
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-10-01 , DOI: 10.1021/acs.jctc.1c00726 Joshua Finkelstein 1 , Justin S Smith 1 , Susan M Mniszewski 2 , Kipton Barros 1 , Christian F A Negre 1 , Emanuel H Rubensson 3 , Anders M N Niklasson 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-10-01 , DOI: 10.1021/acs.jctc.1c00726 Joshua Finkelstein 1 , Justin S Smith 1 , Susan M Mniszewski 2 , Kipton Barros 1 , Christian F A Negre 1 , Emanuel H Rubensson 3 , Anders M N Niklasson 1
Affiliation
|
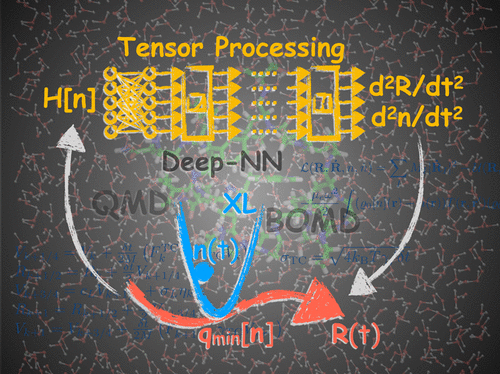
Tensor cores, along with tensor processing units, represent a new form of hardware acceleration specifically designed for deep neural network calculations in artificial intelligence applications. Tensor cores provide extraordinary computational speed and energy efficiency but with the caveat that they were designed for tensor contractions (matrix–matrix multiplications) using only low-precision floating-point operations. Despite this perceived limitation, we demonstrate how tensor cores can be applied with high efficiency to the challenging and numerically sensitive problem of quantum-based Born–Oppenheimer molecular dynamics, which requires highly accurate electronic structure optimizations and conservative force evaluations. The interatomic forces are calculated on-the-fly from an electronic structure that is obtained from a generalized deep neural network, where the computational structure naturally takes advantage of the exceptional processing power of the tensor cores and allows for high performance in excess of 100 Tflops on a single Nvidia A100 GPU. Stable molecular dynamics trajectories are generated using the framework of extended Lagrangian Born–Oppenheimer molecular dynamics, which combines computational efficiency with long-term stability, even when using approximate charge relaxations and force evaluations that are limited in accuracy by the numerically noisy conditions caused by the low-precision tensor core floating-point operations. A canonical ensemble simulation scheme is also presented, where the additional numerical noise in the calculated forces is absorbed into a Langevin-like dynamics.
中文翻译:

使用张量核进行基于量子的分子动力学模拟
张量核心以及张量处理单元代表了一种新形式的硬件加速,专为人工智能应用中的深度神经网络计算而设计。张量核心提供了非凡的计算速度和能源效率,但需要注意的是,它们是为仅使用低精度浮点运算的张量收缩(矩阵-矩阵乘法)而设计的。尽管存在这种感知限制,但我们展示了如何高效地将张量核应用于基于量子的 Born-Oppenheimer 分子动力学的具有挑战性和数值敏感的问题,这需要高度准确的电子结构优化和保守力评估。原子间力是根据从广义深度神经网络获得的电子结构实时计算的,其中计算结构自然利用张量核心的卓越处理能力,并允许超过 100 Tflops 的高性能在单个 Nvidia A100 GPU 上。稳定的分子动力学轨迹是使用扩展的拉格朗日波恩-奥本海默分子动力学框架生成的,该框架将计算效率与长期稳定性相结合,即使在使用近似电荷弛豫和力评估时,其准确性也会受到数值噪声条件的限制。低精度张量核心浮点运算。还提出了一个典型的集成仿真方案,
更新日期:2021-10-12
中文翻译:
使用张量核进行基于量子的分子动力学模拟
张量核心以及张量处理单元代表了一种新形式的硬件加速,专为人工智能应用中的深度神经网络计算而设计。张量核心提供了非凡的计算速度和能源效率,但需要注意的是,它们是为仅使用低精度浮点运算的张量收缩(矩阵-矩阵乘法)而设计的。尽管存在这种感知限制,但我们展示了如何高效地将张量核应用于基于量子的 Born-Oppenheimer 分子动力学的具有挑战性和数值敏感的问题,这需要高度准确的电子结构优化和保守力评估。原子间力是根据从广义深度神经网络获得的电子结构实时计算的,其中计算结构自然利用张量核心的卓越处理能力,并允许超过 100 Tflops 的高性能在单个 Nvidia A100 GPU 上。稳定的分子动力学轨迹是使用扩展的拉格朗日波恩-奥本海默分子动力学框架生成的,该框架将计算效率与长期稳定性相结合,即使在使用近似电荷弛豫和力评估时,其准确性也会受到数值噪声条件的限制。低精度张量核心浮点运算。还提出了一个典型的集成仿真方案,


























 京公网安备 11010802027423号
京公网安备 11010802027423号