当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Ab Initio Direct Dynamics
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2021-09-30 , DOI: 10.1021/acs.accounts.1c00390 H Bernhard Schlegel 1
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2021-09-30 , DOI: 10.1021/acs.accounts.1c00390 H Bernhard Schlegel 1
Affiliation
|
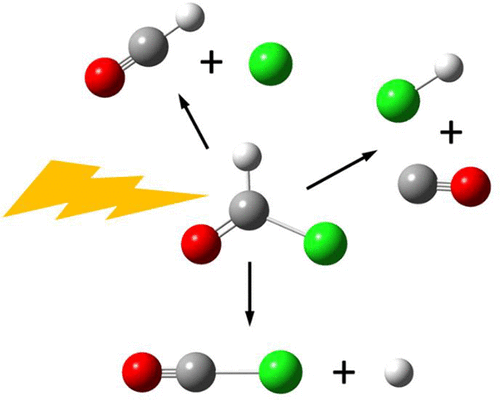
The reactivity and dynamics of molecular systems can be explored computationally by classical trajectory calculations. The traditional approach involves fitting a functional form of a potential energy surface (PES) to the energies from a large number of electronic structure calculations and then integrating numerous trajectories on this fitted PES to model the molecular dynamics. The ever-decreasing cost of computing and continuing advances in computational chemistry software have made it possible to use electronic structure calculations directly in molecular dynamics simulations without first having to construct a fitted PES. In this “on-the-fly” approach, every time the energy and its derivatives are needed for the integration of the equations of motion, they are obtained directly from quantum chemical calculations. This approach started to become practical in the mid-1990s as a result of increased availability of inexpensive computer resources and improved computational chemistry software. The application of direct dynamics calculations has grown rapidly over the last 25 years and would require a lengthy review article. The present Account is limited to some of our contributions to methods development and various applications. To improve the efficiency of direct dynamics calculations, we developed a Hessian-based predictor-corrector algorithm for integrating classical trajectories. Hessian updating made this even more efficient. This approach was also used to improve algorithms for following the steepest descent reaction paths. For larger molecular systems, we developed an extended Lagrangian approach in which the electronic structure is propagated along with the molecular structure. Strong field chemistry is a rapidly growing area, and to improve the accuracy of molecular dynamics in intense laser fields, we included the time-varying electric field in a novel predictor-corrector trajectory integration algorithm. Since intense laser fields can excite and ionize molecules, we extended our studies to include electron dynamics. Specifically, we developed code for time-dependent configuration interaction electron dynamics to simulate strong field ionization by intense laser pulses. Our initial application of ab initio direct dynamics in 1994 was to CH2O → H2 + CO; the calculated vibrational distributions in the products were in very good agreement with experiment. In the intervening years, we have used direct dynamics to explore energy partitioning in various dissociation reactions, unimolecular dissociations yielding three fragments, reactions with branching after the transition state, nonstatistical dynamics of chemically activated molecules, dynamics of molecular fragmentation by intense infrared laser pulses, selective activation of specific dissociation channels by aligned intense infrared laser fields, angular dependence of strong field ionization, and simulation of sequential double ionization.
中文翻译:

Ab Initio 直接动力学
分子系统的反应性和动力学可以通过经典轨迹计算进行计算探索。传统方法涉及将势能面 (PES) 的函数形式与来自大量电子结构计算的能量进行拟合,然后在此拟合的 PES 上整合众多轨迹以模拟分子动力学。不断降低的计算成本和计算化学软件的不断进步使得直接在分子动力学模拟中使用电子结构计算成为可能,而无需首先构建合适的 PES。在这种“即时”方法中,每次需要能量及其导数来积分运动方程时,它们都是直接从量子化学计算中获得的。由于廉价计算机资源的可用性增加和计算化学软件的改进,这种方法在 1990 年代中期开始变得实用。直接动力学计算的应用在过去 25 年中发展迅速,需要一篇冗长的评论文章。目前的帐户仅限于我们对方法开发和各种应用的一些贡献。为了提高直接动力学计算的效率,我们开发了一种基于 Hessian 的预测校正算法,用于整合经典轨迹。Hessian 更新使这更加有效。该方法还用于改进遵循最速下降反应路径的算法。对于较大的分子系统,我们开发了一种扩展的拉格朗日方法,其中电子结构与分子结构一起传播。强场化学是一个快速发展的领域,为了提高强激光场中分子动力学的准确性,我们将时变电场纳入了一种新颖的预测器-校正器轨迹集成算法。由于强激光场可以激发和电离分子,我们将研究扩展到包括电子动力学。具体来说,我们开发了时间相关配置相互作用电子动力学的代码,以模拟强激光脉冲引起的强场电离。我们在 1994 年首次将 ab initio 直接动力学应用于 CH 我们将时变电场包含在一种新颖的预测器-校正器轨迹集成算法中。由于强激光场可以激发和电离分子,我们将研究扩展到包括电子动力学。具体来说,我们开发了时间相关配置相互作用电子动力学的代码,以模拟强激光脉冲引起的强场电离。我们在 1994 年首次将 ab initio 直接动力学应用于 CH 我们将时变电场包含在一种新颖的预测器-校正器轨迹集成算法中。由于强激光场可以激发和电离分子,我们将研究扩展到包括电子动力学。具体来说,我们开发了时间相关配置相互作用电子动力学的代码,以模拟强激光脉冲引起的强场电离。我们在 1994 年首次将 ab initio 直接动力学应用于 CH2 O → H 2 + CO;产品中计算出的振动分布与实验非常吻合。在此期间,我们使用直接动力学探索了各种解离反应中的能量分配、产生三个片段的单分子解离、过渡态后分支的反应、化学活化分子的非统计动力学、强红外激光脉冲引起的分子断裂动力学、通过对准的强红外激光场选择性激活特定解离通道,强场电离的角度依赖性以及连续双电离的模拟。
更新日期:2021-10-19
中文翻译:
Ab Initio 直接动力学
分子系统的反应性和动力学可以通过经典轨迹计算进行计算探索。传统方法涉及将势能面 (PES) 的函数形式与来自大量电子结构计算的能量进行拟合,然后在此拟合的 PES 上整合众多轨迹以模拟分子动力学。不断降低的计算成本和计算化学软件的不断进步使得直接在分子动力学模拟中使用电子结构计算成为可能,而无需首先构建合适的 PES。在这种“即时”方法中,每次需要能量及其导数来积分运动方程时,它们都是直接从量子化学计算中获得的。由于廉价计算机资源的可用性增加和计算化学软件的改进,这种方法在 1990 年代中期开始变得实用。直接动力学计算的应用在过去 25 年中发展迅速,需要一篇冗长的评论文章。目前的帐户仅限于我们对方法开发和各种应用的一些贡献。为了提高直接动力学计算的效率,我们开发了一种基于 Hessian 的预测校正算法,用于整合经典轨迹。Hessian 更新使这更加有效。该方法还用于改进遵循最速下降反应路径的算法。对于较大的分子系统,我们开发了一种扩展的拉格朗日方法,其中电子结构与分子结构一起传播。强场化学是一个快速发展的领域,为了提高强激光场中分子动力学的准确性,我们将时变电场纳入了一种新颖的预测器-校正器轨迹集成算法。由于强激光场可以激发和电离分子,我们将研究扩展到包括电子动力学。具体来说,我们开发了时间相关配置相互作用电子动力学的代码,以模拟强激光脉冲引起的强场电离。我们在 1994 年首次将 ab initio 直接动力学应用于 CH 我们将时变电场包含在一种新颖的预测器-校正器轨迹集成算法中。由于强激光场可以激发和电离分子,我们将研究扩展到包括电子动力学。具体来说,我们开发了时间相关配置相互作用电子动力学的代码,以模拟强激光脉冲引起的强场电离。我们在 1994 年首次将 ab initio 直接动力学应用于 CH 我们将时变电场包含在一种新颖的预测器-校正器轨迹集成算法中。由于强激光场可以激发和电离分子,我们将研究扩展到包括电子动力学。具体来说,我们开发了时间相关配置相互作用电子动力学的代码,以模拟强激光脉冲引起的强场电离。我们在 1994 年首次将 ab initio 直接动力学应用于 CH2 O → H 2 + CO;产品中计算出的振动分布与实验非常吻合。在此期间,我们使用直接动力学探索了各种解离反应中的能量分配、产生三个片段的单分子解离、过渡态后分支的反应、化学活化分子的非统计动力学、强红外激光脉冲引起的分子断裂动力学、通过对准的强红外激光场选择性激活特定解离通道,强场电离的角度依赖性以及连续双电离的模拟。

























 京公网安备 11010802027423号
京公网安备 11010802027423号