当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Multiscale Modeling of Electronic Spectra Including Nuclear Quantum Effects
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-28 , DOI: 10.1021/acs.jctc.1c00531 Péter P Fehér 1 , Ádám Madarász 1 , András Stirling 1, 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-28 , DOI: 10.1021/acs.jctc.1c00531 Péter P Fehér 1 , Ádám Madarász 1 , András Stirling 1, 2
Affiliation
|
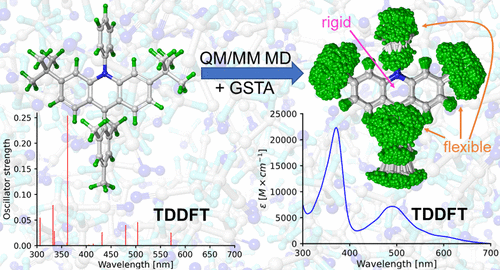
Theoretical prediction of electronic absorption spectra without input from experiments is no easy feat, as it requires addressing all of the factors that affect line shapes. In practice, however, the methodologies are limited to treat these ingredients only to a certain extent. Here, we present a multiscale protocol that addresses the temperature, solvent, and nuclear quantum effects as well as anharmonicity and the reconstruction of the final spectra from individual transitions. First, quantum mechanics/molecular mechanics (QM/MM) molecular dynamics is conducted to obtain trajectories of solute–solvent configurations, from which the corresponding quantum-corrected ensembles are generated through the generalized smoothed trajectory analysis (GSTA). The optical spectra of the ensembles are then produced by calculating vertical transitions using time-dependent density-functional theory (TDDFT) with implicit solvation. To obtain the final spectral shapes, the stick spectra from TDDFT are convoluted with Gaussian kernels where the half-widths are determined by a statistically motivated strategy. We have tested our method by calculating the UV–vis spectra of a recently discovered acridine photocatalyst in two redox states. Vibronic progressions and broadenings due to the finite lifetime of the excited states are not included in the methodology yet. Nuclear quantization affects the relative peak intensities and widths, which is necessary to reproduce the experimental spectrum. We have also found that using only the optimized geometry of each molecule works surprisingly well if a proper empirical broadening factor is applied. This is explained by the rigidity of the conjugated chromophore moieties of the selected molecules, which are mainly responsible for the excitations in the spectra. In contrast, we have also shown that other parts of the molecules are flexible enough to feature anharmonicities that impair the use of other techniques such as Wigner sampling.
中文翻译:

包括核量子效应在内的电子光谱的多尺度建模
在没有实验输入的情况下对电子吸收光谱进行理论预测并非易事,因为它需要解决影响线条形状的所有因素。然而,在实践中,这些方法仅限于在一定程度上处理这些成分。在这里,我们提出了一个多尺度协议,该协议解决了温度、溶剂和核量子效应以及非谐性和从单个跃迁重建最终光谱的问题。首先,进行量子力学/分子力学 (QM/MM) 分子动力学以获得溶质 - 溶剂配置的轨迹,从中通过广义平滑轨迹分析 (GSTA) 生成相应的量子校正系综。然后通过使用具有隐式溶剂化的时间相关密度泛函理论 (TDDFT) 计算垂直跃迁来产生集合的光谱。为了获得最终的光谱形状,来自 TDDFT 的棒状光谱与高斯核进行卷积,其中半宽由统计激励策略确定。我们通过计算最近发现的两种氧化还原状态的吖啶光催化剂的紫外-可见光谱来测试我们的方法。由于激发态的有限寿命而导致的振动级数和展宽尚未包含在该方法中。核量子化影响相对峰值强度和宽度,这是重现实验光谱所必需的。我们还发现,如果应用适当的经验加宽因子,则仅使用每个分子的优化几何形状效果出奇地好。这是通过所选分子的共轭发色团部分的刚性来解释的,它们主要负责光谱中的激发。相比之下,我们还表明,分子的其他部分足够灵活,具有不协调性,会损害其他技术(如 Wigner 采样)的使用。
更新日期:2021-10-12
中文翻译:
包括核量子效应在内的电子光谱的多尺度建模
在没有实验输入的情况下对电子吸收光谱进行理论预测并非易事,因为它需要解决影响线条形状的所有因素。然而,在实践中,这些方法仅限于在一定程度上处理这些成分。在这里,我们提出了一个多尺度协议,该协议解决了温度、溶剂和核量子效应以及非谐性和从单个跃迁重建最终光谱的问题。首先,进行量子力学/分子力学 (QM/MM) 分子动力学以获得溶质 - 溶剂配置的轨迹,从中通过广义平滑轨迹分析 (GSTA) 生成相应的量子校正系综。然后通过使用具有隐式溶剂化的时间相关密度泛函理论 (TDDFT) 计算垂直跃迁来产生集合的光谱。为了获得最终的光谱形状,来自 TDDFT 的棒状光谱与高斯核进行卷积,其中半宽由统计激励策略确定。我们通过计算最近发现的两种氧化还原状态的吖啶光催化剂的紫外-可见光谱来测试我们的方法。由于激发态的有限寿命而导致的振动级数和展宽尚未包含在该方法中。核量子化影响相对峰值强度和宽度,这是重现实验光谱所必需的。我们还发现,如果应用适当的经验加宽因子,则仅使用每个分子的优化几何形状效果出奇地好。这是通过所选分子的共轭发色团部分的刚性来解释的,它们主要负责光谱中的激发。相比之下,我们还表明,分子的其他部分足够灵活,具有不协调性,会损害其他技术(如 Wigner 采样)的使用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号