Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2021-09-28 , DOI: 10.1016/j.csbj.2021.09.031 Zehui Yu 1, 2, 3 , Wenjie Zhang 1, 4 , Huancheng Fu 5 , Xiaoxia Zou 6 , Mingde Zhao 1 , Sicheng Liang 2 , Congwei Gu 1 , Qian Yang 1 , Manli He 1 , Qihai Xiao 1 , Wudian Xiao 1 , Lvqin He 1 , Muhan Lü 2
|
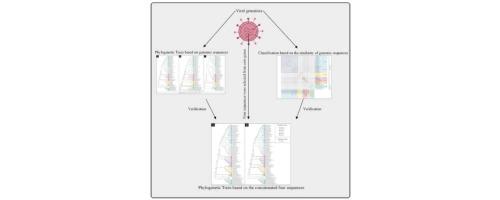
The members of the Poxviridae family are globally distributed all over the world and can cause infectious diseases. Although genome sequences are publicly available for representative isolates of all genera, studies on the criteria for genome-based classification within the Poxviridae family have rarely been reported. In our study, 60 Poxviridae genomes were re-annotated using Prokka. By using BLAST filtration and MCScanX, synteny and similarity of whole genomic amino acid sequences were visualized. According to the analysis pattern, the Chordopoxvirinae and Entomopoxvirinae subfamilies can be subdivided into five and two categories respectively, which is consistent with the phylogenetic tree constructed based on whole genomic amino acid sequences and Poxvirus core genes. Finally, four genes (Early transcription factor, DNA-directed RNA polymerase, RNA polymerase-associated transcription-specificity factor and DNA-dependent RNA polymerase) were selected from Poxvirus core genes by substitution saturation analysis and phylogenetic tree verification. Phylogenetic trees constructed based on single gene and concatenated sequences of the four selected genes showed that the classification of subgroups was consistent with the phylogenetic trees based on genome. Conclusion: a new method based on the similarity of whole genomic amino acid sequences was proposed for Poxviridae taxon demarcation, and the use of the four selected qualified genes will help make phylogenic identification of newly discovered Poxviridae isolates more convenient and accurate.
中文翻译:
痘病毒科基因组分析及系统发育合格基因序列探索
痘病毒科的成员全球分布于世界各地,可引起传染病。尽管所有属的代表性分离株的基因组序列都是公开可用的,但关于痘病毒科内基于基因组的分类标准的研究却鲜有报道。在我们的研究中,使用 Prokka 重新注释了60 个痘病毒科基因组。通过使用 BLAST 过滤和 MCScanX,可视化整个基因组氨基酸序列的同线性和相似性。根据分析模式,脊痘病毒亚科和昆虫痘病毒亚科亚科可分别细分为五个和两个类别,这与基于全基因组氨基酸序列和痘病毒核心基因构建的系统发育树一致。最后,通过替换饱和度分析和系统发育树验证,从痘病毒核心基因中选出了四个基因(早期转录因子、DNA 导向的 RNA 聚合酶、RNA 聚合酶相关的转录特异性因子和 DNA 依赖性 RNA 聚合酶)。基于单个基因和四个选定基因的串联序列构建的系统发育树表明,亚群的分类与基于基因组的系统发育树一致。结论:提出了一种基于全基因组氨基酸序列相似性的痘病毒检测新方法分类群划分,并使用四个选定的合格基因将有助于使新发现的痘病毒分离株的系统发育鉴定更加方便和准确。

























 京公网安备 11010802027423号
京公网安备 11010802027423号