当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Absorption Spectra of Solids from Periodic Equation-of-Motion Coupled-Cluster Theory
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-24 , DOI: 10.1021/acs.jctc.1c00692 Xiao Wang 1 , Timothy C Berkelbach 1, 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-24 , DOI: 10.1021/acs.jctc.1c00692 Xiao Wang 1 , Timothy C Berkelbach 1, 2
Affiliation
|
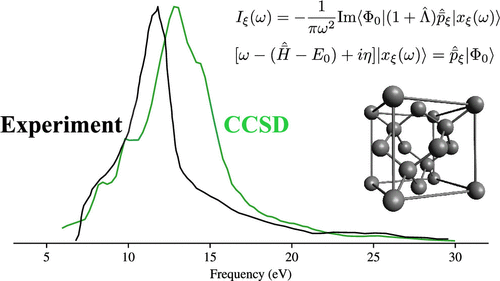
We present ab initio absorption spectra of six three-dimensional semiconductors and insulators calculated using Gaussian-based periodic equation-of-motion coupled-cluster theory with single and double excitations (EOM-CCSD). The spectra are calculated efficiently by solving a system of linear equations at each frequency, giving access to an energy range of tens of electronvolts without explicit enumeration of excited states. We assess the impact of cost-saving approximations associated with Brillouin zone sampling, frozen orbitals, and the partitioned EOM-CCSD approximation. Although our most converged spectra exhibit line shapes that are in good agreement with experimental spectra, they are uniformly shifted to higher energies by about 1 eV, which is not explained by the remaining finite-size errors. We tentatively attribute this discrepancy to a combination of vibrational effects and the remaining electron correlation, i.e., triple excitations and above.
中文翻译:

周期运动方程耦合簇理论中固体的吸收光谱
我们提出了六个三维半导体和绝缘体的从头算吸收光谱,该吸收光谱使用基于高斯的周期运动方程耦合簇理论与单激发和双激发 (EOM-CCSD) 计算。通过在每个频率下求解线性方程组系统,可以有效地计算光谱,从而获得数十电子伏特的能量范围,而无需明确列举激发态。我们评估了与布里渊区采样、冻结轨道和分区 EOM-CCSD 近似相关的成本节约近似的影响。尽管我们最收敛的光谱表现出与实验光谱非常一致的线形,但它们均匀地移动到更高的能量约 1 eV,这无法用剩余的有限尺寸误差来解释。
更新日期:2021-10-12
中文翻译:
周期运动方程耦合簇理论中固体的吸收光谱
我们提出了六个三维半导体和绝缘体的从头算吸收光谱,该吸收光谱使用基于高斯的周期运动方程耦合簇理论与单激发和双激发 (EOM-CCSD) 计算。通过在每个频率下求解线性方程组系统,可以有效地计算光谱,从而获得数十电子伏特的能量范围,而无需明确列举激发态。我们评估了与布里渊区采样、冻结轨道和分区 EOM-CCSD 近似相关的成本节约近似的影响。尽管我们最收敛的光谱表现出与实验光谱非常一致的线形,但它们均匀地移动到更高的能量约 1 eV,这无法用剩余的有限尺寸误差来解释。

























 京公网安备 11010802027423号
京公网安备 11010802027423号