当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Development and Benchmarking of Open Force Field v1.0.0—the Parsley Small-Molecule Force Field
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-22 , DOI: 10.1021/acs.jctc.1c00571 Yudong Qiu 1 , Daniel G A Smith 2 , Simon Boothroyd 3 , Hyesu Jang 1 , David F Hahn 4 , Jeffrey Wagner 5 , Caitlin C Bannan 5, 6 , Trevor Gokey 5 , Victoria T Lim 5 , Chaya D Stern 3 , Andrea Rizzi 3, 7 , Bryon Tjanaka 5 , Gary Tresadern 4 , Xavier Lucas 8 , Michael R Shirts 9 , Michael K Gilson 6 , John D Chodera 3 , Christopher I Bayly 10 , David L Mobley 5 , Lee-Ping Wang 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-22 , DOI: 10.1021/acs.jctc.1c00571 Yudong Qiu 1 , Daniel G A Smith 2 , Simon Boothroyd 3 , Hyesu Jang 1 , David F Hahn 4 , Jeffrey Wagner 5 , Caitlin C Bannan 5, 6 , Trevor Gokey 5 , Victoria T Lim 5 , Chaya D Stern 3 , Andrea Rizzi 3, 7 , Bryon Tjanaka 5 , Gary Tresadern 4 , Xavier Lucas 8 , Michael R Shirts 9 , Michael K Gilson 6 , John D Chodera 3 , Christopher I Bayly 10 , David L Mobley 5 , Lee-Ping Wang 1
Affiliation
|
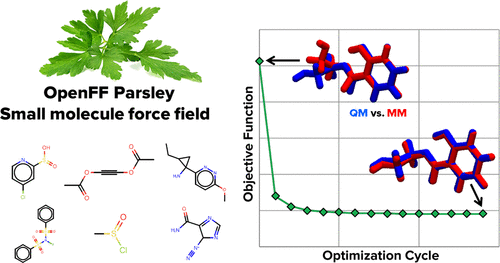
We present a methodology for defining and optimizing a general force field for classical molecular simulations, and we describe its use to derive the Open Force Field 1.0.0 small-molecule force field, codenamed Parsley. Rather than using traditional atom typing, our approach is built on the SMIRKS-native Open Force Field (SMIRNOFF) parameter assignment formalism, which handles increases in the diversity and specificity of the force field definition without needlessly increasing the complexity of the specification. Parameters are optimized with the ForceBalance tool, based on reference quantum chemical data that include torsion potential energy profiles, optimized gas-phase structures, and vibrational frequencies. These quantum reference data are computed and are maintained with QCArchive, an open-source and freely available distributed computing and database software ecosystem. In this initial application of the method, we present essentially a full optimization of all valence parameters and report tests of the resulting force field against compounds and data types outside the training set. These tests show improvements in optimized geometries and conformational energetics and demonstrate that Parsley’s accuracy for liquid properties is similar to that of other general force fields, as is accuracy on binding free energies. We find that this initial Parsley force field affords accuracy similar to that of other general force fields when used to calculate relative binding free energies spanning 199 protein–ligand systems. Additionally, the resulting infrastructure allows us to rapidly optimize an entirely new force field with minimal human intervention.
中文翻译:

Open力场v1.0.0的开发和基准测试——欧芹小分子力场
我们提出了一种定义和优化经典分子模拟通用力场的方法,并描述了其用于推导 Open Force Field 1.0.0 小分子力场(代号为 Parsley)的方法。我们的方法不是使用传统的原子类型,而是建立在 SMIRKS 原生开放力场 (SMIRNOFF) 参数分配形式之上,它可以处理力场定义的多样性和特异性的增加,而不会不必要地增加规范的复杂性。使用 ForceBalance 工具根据参考量子化学数据(包括扭转势能分布、优化的气相结构和振动频率)优化参数。这些量子参考数据通过 QCArchive 进行计算和维护,QCArchive 是一个开源且免费的分布式计算和数据库软件生态系统。在该方法的初始应用中,我们本质上提出了所有价参数的全面优化,并报告了针对训练集之外的化合物和数据类型对所得力场的测试。这些测试显示了优化几何结构和构象能量学的改进,并证明 Parsley 的液体特性准确性与其他一般力场的准确性相似,结合自由能的准确性也是如此。我们发现,当用于计算跨越 199 个蛋白质-配体系统的相对结合自由能时,这个初始欧芹力场提供了与其他一般力场相似的精度。此外,由此产生的基础设施使我们能够以最少的人为干预快速优化全新的力场。
更新日期:2021-10-12
中文翻译:
Open力场v1.0.0的开发和基准测试——欧芹小分子力场
我们提出了一种定义和优化经典分子模拟通用力场的方法,并描述了其用于推导 Open Force Field 1.0.0 小分子力场(代号为 Parsley)的方法。我们的方法不是使用传统的原子类型,而是建立在 SMIRKS 原生开放力场 (SMIRNOFF) 参数分配形式之上,它可以处理力场定义的多样性和特异性的增加,而不会不必要地增加规范的复杂性。使用 ForceBalance 工具根据参考量子化学数据(包括扭转势能分布、优化的气相结构和振动频率)优化参数。这些量子参考数据通过 QCArchive 进行计算和维护,QCArchive 是一个开源且免费的分布式计算和数据库软件生态系统。在该方法的初始应用中,我们本质上提出了所有价参数的全面优化,并报告了针对训练集之外的化合物和数据类型对所得力场的测试。这些测试显示了优化几何结构和构象能量学的改进,并证明 Parsley 的液体特性准确性与其他一般力场的准确性相似,结合自由能的准确性也是如此。我们发现,当用于计算跨越 199 个蛋白质-配体系统的相对结合自由能时,这个初始欧芹力场提供了与其他一般力场相似的精度。此外,由此产生的基础设施使我们能够以最少的人为干预快速优化全新的力场。


























 京公网安备 11010802027423号
京公网安备 11010802027423号