当前位置:
X-MOL 学术
›
Anal. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Probing Universal Protein Dynamics Using Hydrogen–Deuterium Exchange Mass Spectrometry-Derived Residue-Level Gibbs Free Energy
Analytical Chemistry ( IF 7.4 ) Pub Date : 2021-09-15 , DOI: 10.1021/acs.analchem.1c02155 Jochem H Smit 1 , Srinath Krishnamurthy 1 , Bindu Y Srinivasu 1 , Rinky Parakra 1 , Spyridoula Karamanou 1 , Anastassios Economou 1
Analytical Chemistry ( IF 7.4 ) Pub Date : 2021-09-15 , DOI: 10.1021/acs.analchem.1c02155 Jochem H Smit 1 , Srinath Krishnamurthy 1 , Bindu Y Srinivasu 1 , Rinky Parakra 1 , Spyridoula Karamanou 1 , Anastassios Economou 1
Affiliation
|
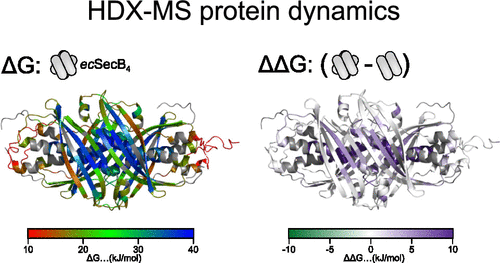
Hydrogen–deuterium exchange mass spectrometry (HDX-MS) is a powerful technique to monitor protein intrinsic dynamics. The technique provides high-resolution information on how protein intrinsic dynamics are altered in response to biological signals, such as ligand binding, oligomerization, or allosteric networks. However, identification, interpretation, and visualization of such events from HDX-MS data sets is challenging as these data sets consist of many individual data points collected across peptides, time points, and experimental conditions. Here, we present PyHDX, an open-source Python package and webserver, that allows the user to batch extract the universal quantity Gibbs free energy at residue levels over multiple protein conditions and homologues. The output is directly visualized on a linear map or 3D structures or is exported as .csv files or PyMOL scripts.
中文翻译:

使用氢-氘交换质谱法衍生的残留水平吉布斯自由能探索通用蛋白质动力学
氢-氘交换质谱 (HDX-MS) 是一种监测蛋白质内在动力学的强大技术。该技术提供了有关蛋白质内在动力学如何响应生物信号(例如配体结合、寡聚化或变构网络)而改变的高分辨率信息。然而,从 HDX-MS 数据集中识别、解释和可视化此类事件具有挑战性,因为这些数据集由跨肽、时间点和实验条件收集的许多单独数据点组成。在这里,我们展示了 PyHDX,一个开源 Python 包和网络服务器,它允许用户在多个蛋白质条件和同系物的残留水平上批量提取通用量吉布斯自由能。输出直接在线性地图或 3D 结构上可视化,或导出为 .
更新日期:2021-09-28
中文翻译:
使用氢-氘交换质谱法衍生的残留水平吉布斯自由能探索通用蛋白质动力学
氢-氘交换质谱 (HDX-MS) 是一种监测蛋白质内在动力学的强大技术。该技术提供了有关蛋白质内在动力学如何响应生物信号(例如配体结合、寡聚化或变构网络)而改变的高分辨率信息。然而,从 HDX-MS 数据集中识别、解释和可视化此类事件具有挑战性,因为这些数据集由跨肽、时间点和实验条件收集的许多单独数据点组成。在这里,我们展示了 PyHDX,一个开源 Python 包和网络服务器,它允许用户在多个蛋白质条件和同系物的残留水平上批量提取通用量吉布斯自由能。输出直接在线性地图或 3D 结构上可视化,或导出为 .

























 京公网安备 11010802027423号
京公网安备 11010802027423号