Computational Materials Science ( IF 3.3 ) Pub Date : 2021-09-14 , DOI: 10.1016/j.commatsci.2021.110836 Spencer Wyant 1 , Andrew Rohskopf 2 , Asegun Henry 2
|
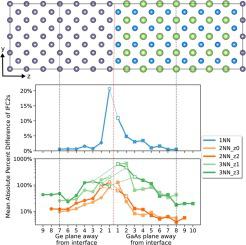
Molecular dynamics simulations provide a versatile framework to study interfacial heat transport, but their accuracy remains limited by the accuracy of available interatomic potentials. Past researchers relied on simple analytic potentials and the use of mixing rules to model interface systems, but with minimal justification for their use. Contemporary researchers have access to highly flexible machine learned interatomic potentials (MLIPs), yet these remain understudied within this problem domain. In either case, efforts are needed to rigorously assess and validate interatomic potentials prior to their use in MD simulations of interfacial heat transport. Here, we pursue these efforts while developing interatomic potentials for a model Ge/GaAs system. We first assess the quality of ab initio harmonic force constants (IFC2s) obtained from the interface structure that is used to generate fitting data for MLIPs. We show that these force constants can converge to near bulk-like values within 1 nm away from the interface while also exhibiting a complex relationship across the interface, which likely precludes any successful application of mixing rules. Subsequently, we develop two different MLIPs to model Ge/GaAs using the linear spectral neighborhood analysis potential (SNAP) functional form: one standard SNAP fit to total forces, and another hybrid SNAP fit to anharmonic force components and combined with a harmonic Taylor expansion potential. Each potential is evaluated with respect to bulk thermal properties, interface IFC2s, and stability considerations, providing a valuable comparative analysis that can guide future work.
中文翻译:
用于模拟 Ge/GaAs 界面热传输的机器学习原子间势
分子动力学模拟为研究界面热传递提供了一个通用框架,但其准确性仍然受到可用原子间势的准确性的限制。过去的研究人员依靠简单的分析势和使用混合规则来模拟接口系统,但使用它们的理由很少。当代研究人员可以使用高度灵活的机器学习原子间势 (MLIP),但这些在这个问题领域内仍未得到充分研究。在任何一种情况下,在将原子间势用于界面热传输的 MD 模拟之前,都需要努力严格评估和验证原子间势。在这里,我们在为模型 Ge/GaAs 系统开发原子间势的同时进行这些努力。我们首先评估从头开始的质量从用于生成 MLIP 拟合数据的界面结构获得的谐波力常数 (IFC2)。我们表明,这些力常数可以收敛到距界面 1 nm 以内的接近体积的值,同时还表现出跨界面的复杂关系,这可能会妨碍混合规则的任何成功应用。随后,我们开发了两种不同的 MLIP 来使用线性谱邻域分析势 (SNAP) 函数形式对 Ge/GaAs 进行建模:一种标准 SNAP 拟合合力,另一种混合 SNAP 拟合非谐力分量并结合谐波泰勒展开势. 每种潜力都根据体热特性、界面 IFC2 和稳定性考虑进行评估,提供有价值的比较分析,可以指导未来的工作。
























 京公网安备 11010802027423号
京公网安备 11010802027423号