Current Bioinformatics ( IF 4 ) Pub Date : 2021-06-30 , DOI: 10.2174/1574893616666210430133846 Saud Alguwaizani 1 , Shulei Ren 1 , De-Shuang Huang 2 , Kyungsook Han 1
|
Aim: Both bacterial infection and viral infection involve a large number of protein-protein interactions (PPIs) between a pathogen and its target host.
Background: So far, many computational methods have focused on predicting PPIs within the same species rather than PPIs across different species.
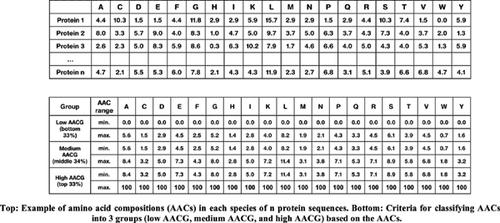
Methods: From the extensive analysis of PPIs between Yersinia pestis bacteria and humans, we recently discovered an interesting relation; a linear relation between amino acid composition and sequence length was observed in many proteins involved in PPIs. We have built a support vector machine (SVM) model, which predicts PPIs between human and bacteria using two feature types derived from the relation. The two feature types used in the SVM are the amino acid composition group (AACG) and the difference in amino acid composition between host and pathogen proteins.
Results: The SVM model achieved high performance in predicting bacteria-human PPIs. The model showed an accuracy of 96%, sensitivity of 94%, and specificity of 98% in predicting PPIs between humans and Yersinia pestis, in which there is a strong relation between amino acid composition and sequence length. The SVM model was also tested in predicting PPIs between human and viruses, which include Ebola, HCV, and SARS-CoV-2, and showed a good performance.
Conclusion: The feature types identified in our study are simple yet powerful in predicting pathogenhuman PPIs. Although preliminary, our method will be useful for finding unknown target host proteins or pathogen proteins and designing in vitro or in vivo experiments.
中文翻译:
基于序列长度和氨基酸组成之间的关系预测病原体和人类蛋白质之间的相互作用
目的:细菌感染和病毒感染都涉及病原体与其目标宿主之间的大量蛋白质-蛋白质相互作用 (PPI)。
背景:到目前为止,许多计算方法都侧重于预测同一物种内的 PPI,而不是预测不同物种之间的 PPI。
方法:通过对鼠疫杆菌与人类之间 PPI 的广泛分析,我们最近发现了一个有趣的关系;在参与 PPI 的许多蛋白质中观察到氨基酸组成和序列长度之间的线性关系。我们建立了一个支持向量机 (SVM) 模型,该模型使用从关系派生的两种特征类型来预测人类和细菌之间的 PPI。SVM 中使用的两种特征类型是氨基酸组成组 (AACG) 和宿主和病原体蛋白质之间氨基酸组成的差异。
结果:SVM 模型在预测细菌-人类 PPI 方面取得了高性能。该模型在预测人类与鼠疫耶尔森菌之间的 PPI 方面的准确度为 96%,灵敏度为 94%,特异性为 98%,其中氨基酸组成与序列长度之间存在很强的相关性。SVM 模型在预测人类与病毒(包括埃博拉、HCV 和 SARS-CoV-2)之间的 PPI 方面也进行了测试,并显示出良好的性能。
结论:我们研究中确定的特征类型在预测病原体人类 PPI 方面既简单又有效。虽然是初步的,但我们的方法将有助于寻找未知的目标宿主蛋白或病原体蛋白以及设计体外或体内实验。

























 京公网安备 11010802027423号
京公网安备 11010802027423号