当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Machine learning to tame divergent density functional approximations: a new path to consensus materials design principles
Chemical Science ( IF 8.4 ) Pub Date : 2021-09-02 , DOI: 10.1039/d1sc03701c Chenru Duan 1, 2 , Shuxin Chen 1, 2 , Michael G Taylor 1 , Fang Liu 1 , Heather J Kulik 1
Chemical Science ( IF 8.4 ) Pub Date : 2021-09-02 , DOI: 10.1039/d1sc03701c Chenru Duan 1, 2 , Shuxin Chen 1, 2 , Michael G Taylor 1 , Fang Liu 1 , Heather J Kulik 1
Affiliation
|
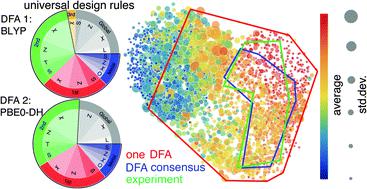
Virtual high-throughput screening (VHTS) with density functional theory (DFT) and machine-learning (ML)-acceleration is essential in rapid materials discovery. By necessity, efficient DFT-based workflows are carried out with a single density functional approximation (DFA). Nevertheless, properties evaluated with different DFAs can be expected to disagree for cases with challenging electronic structure (e.g., open-shell transition-metal complexes, TMCs) for which rapid screening is most needed and accurate benchmarks are often unavailable. To quantify the effect of DFA bias, we introduce an approach to rapidly obtain property predictions from 23 representative DFAs spanning multiple families, “rungs” (e.g., semi-local to double hybrid) and basis sets on over 2000 TMCs. Although computed property values (e.g., spin state splitting and frontier orbital gap) differ by DFA, high linear correlations persist across all DFAs. We train independent ML models for each DFA and observe convergent trends in feature importance, providing DFA-invariant, universal design rules. We devise a strategy to train artificial neural network (ANN) models informed by all 23 DFAs and use them to predict properties (e.g., spin-splitting energy) of over 187k TMCs. By requiring consensus of the ANN-predicted DFA properties, we improve correspondence of computational lead compounds with literature-mined, experimental compounds over the typically employed single-DFA approach.
中文翻译:

机器学习驯服发散密度函数近似:共识材料设计原则的新途径
具有密度泛函理论 (DFT) 和机器学习 (ML) 加速的虚拟高通量筛选 (VHTS) 对于快速发现材料至关重要。必须使用单密度函数近似 (DFA) 执行基于 DFT 的高效工作流程。然而,对于具有挑战性的电子结构(例如,开壳过渡金属配合物,TMCs)的情况,用不同的 DFA 评估的特性可能会不一致,因为这些情况最需要快速筛选并且通常无法获得准确的基准。为了量化 DFA 偏差的影响,我们引入了一种方法来快速从 23 个跨多个族、“梯级”(例如、半本地到双混合)和基于 2000 多个 TMC 的基组。尽管计算的属性值(例如,自旋状态分裂和前沿轨道间隙)因 DFA 而异,但所有 DFA 之间仍然存在高线性相关性。我们为每个 DFA 训练独立的 ML 模型并观察特征重要性的收敛趋势,提供 DFA 不变的通用设计规则。我们设计了一种策略来训练由所有 23 个 DFA 提供信息的人工神经网络 (ANN) 模型,并使用它们来预测超过 187k TMC 的特性(例如,自旋分裂能量)。通过要求对 ANN 预测的 DFA 属性达成共识,我们改进了计算先导化合物与文献挖掘的实验化合物的对应关系,而不是通常采用的单 DFA 方法。
更新日期:2021-09-13
中文翻译:
机器学习驯服发散密度函数近似:共识材料设计原则的新途径
具有密度泛函理论 (DFT) 和机器学习 (ML) 加速的虚拟高通量筛选 (VHTS) 对于快速发现材料至关重要。必须使用单密度函数近似 (DFA) 执行基于 DFT 的高效工作流程。然而,对于具有挑战性的电子结构(例如,开壳过渡金属配合物,TMCs)的情况,用不同的 DFA 评估的特性可能会不一致,因为这些情况最需要快速筛选并且通常无法获得准确的基准。为了量化 DFA 偏差的影响,我们引入了一种方法来快速从 23 个跨多个族、“梯级”(例如、半本地到双混合)和基于 2000 多个 TMC 的基组。尽管计算的属性值(例如,自旋状态分裂和前沿轨道间隙)因 DFA 而异,但所有 DFA 之间仍然存在高线性相关性。我们为每个 DFA 训练独立的 ML 模型并观察特征重要性的收敛趋势,提供 DFA 不变的通用设计规则。我们设计了一种策略来训练由所有 23 个 DFA 提供信息的人工神经网络 (ANN) 模型,并使用它们来预测超过 187k TMC 的特性(例如,自旋分裂能量)。通过要求对 ANN 预测的 DFA 属性达成共识,我们改进了计算先导化合物与文献挖掘的实验化合物的对应关系,而不是通常采用的单 DFA 方法。

























 京公网安备 11010802027423号
京公网安备 11010802027423号