当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Effect of the Force Field on Molecular Dynamics Simulations of the Multidrug Efflux Protein P-Glycoprotein
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-10 , DOI: 10.1021/acs.jctc.1c00414 Lily Wang 1 , Megan L O'Mara 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-10 , DOI: 10.1021/acs.jctc.1c00414 Lily Wang 1 , Megan L O'Mara 1
Affiliation
|
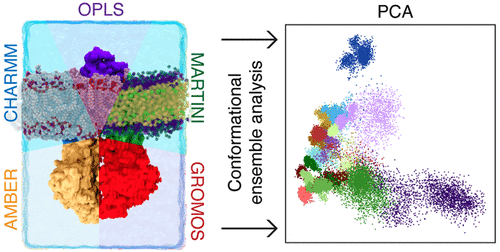
Molecular dynamics (MD) simulations have been used extensively to study P-glycoprotein (P-gp), a flexible multidrug transporter that is a key player in the development of multidrug resistance to chemotherapeutics. A substantial body of literature has grown from simulation studies that have employed various simulation conditions and parameters, including AMBER, CHARMM, OPLS, GROMOS, and coarse-grained force fields, drawing conclusions from simulations spanning hundreds of nanoseconds. Each force field is typically parametrized and validated on different data and observables, usually of small molecules and peptides; there have been few comparisons of force field performance on large protein–membrane systems. Here we compare the conformational ensembles of P-gp embedded in a POPC/cholesterol bilayer generated over 500 ns of replicate simulation with five force fields from popular biomolecular families: AMBER 99SB-ILDN, CHARMM 36, OPLS-AA/L, GROMOS 54A7, and MARTINI. We find considerable differences among the ensembles with little conformational overlap, although they correspond to similar extents to structural data obtained from electron paramagnetic resonance and cross-linking studies. Moreover, each trajectory was still sampling new conformations at a high rate after 500 ns of simulation, suggesting the need for more sampling. This work highlights the need to consider known limitations of the force field used (e.g., biases toward certain secondary structures) and the simulation itself (e.g., whether sufficient sampling has been achieved) when interpreting accumulated results of simulation studies of P-gp and other transport proteins.
中文翻译:

力场对多药外排蛋白 P-糖蛋白分子动力学模拟的影响
分子动力学 (MD) 模拟已被广泛用于研究 P-糖蛋白 (P-gp),这是一种灵活的多药转运蛋白,是发展对化疗药物的多药耐药性的关键参与者。大量文献来自模拟研究,这些模拟研究采用了各种模拟条件和参数,包括 AMBER、CHARMM、OPLS、GROMOS 和粗粒度力场,从跨越数百纳秒的模拟中得出结论。每个力场通常在不同的数据和可观察的数据(通常是小分子和肽)上进行参数化和验证;很少有大型蛋白质膜系统上的力场性能比较。在这里,我们比较了嵌入在 POPC/胆固醇双层中的 P-gp 的构象集合,生成了超过 500 ns 的复制模拟和来自流行生物分子家族的五个力场:AMBER 99SB-ILDN、CHARMM 36、OPLS-AA/L、GROMOS 54A7、和马丁尼。我们发现几乎没有构象重叠的集合之间存在相当大的差异,尽管它们对应于从电子顺磁共振和交联研究获得的结构数据的相似程度。此外,在 500 ns 的模拟之后,每个轨迹仍在高速采样新构象,这表明需要更多采样。这项工作强调需要考虑使用的力场的已知限制(例如,偏向某些二级结构)和模拟本身(例如,
更新日期:2021-10-12
中文翻译:
力场对多药外排蛋白 P-糖蛋白分子动力学模拟的影响
分子动力学 (MD) 模拟已被广泛用于研究 P-糖蛋白 (P-gp),这是一种灵活的多药转运蛋白,是发展对化疗药物的多药耐药性的关键参与者。大量文献来自模拟研究,这些模拟研究采用了各种模拟条件和参数,包括 AMBER、CHARMM、OPLS、GROMOS 和粗粒度力场,从跨越数百纳秒的模拟中得出结论。每个力场通常在不同的数据和可观察的数据(通常是小分子和肽)上进行参数化和验证;很少有大型蛋白质膜系统上的力场性能比较。在这里,我们比较了嵌入在 POPC/胆固醇双层中的 P-gp 的构象集合,生成了超过 500 ns 的复制模拟和来自流行生物分子家族的五个力场:AMBER 99SB-ILDN、CHARMM 36、OPLS-AA/L、GROMOS 54A7、和马丁尼。我们发现几乎没有构象重叠的集合之间存在相当大的差异,尽管它们对应于从电子顺磁共振和交联研究获得的结构数据的相似程度。此外,在 500 ns 的模拟之后,每个轨迹仍在高速采样新构象,这表明需要更多采样。这项工作强调需要考虑使用的力场的已知限制(例如,偏向某些二级结构)和模拟本身(例如,


























 京公网安备 11010802027423号
京公网安备 11010802027423号