当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
On the Origin of the Different Reversible Characters of Salinosporamide A and Homosalinosporamide A in the Covalent Inhibition of the Human 20S Proteasome
ACS Catalysis ( IF 12.9 ) Pub Date : 2021-09-08 , DOI: 10.1021/acscatal.1c02614 Natalia Serrano-Aparicio 1 , Vicent Moliner 1 , Katarzyna Świderek 1
ACS Catalysis ( IF 12.9 ) Pub Date : 2021-09-08 , DOI: 10.1021/acscatal.1c02614 Natalia Serrano-Aparicio 1 , Vicent Moliner 1 , Katarzyna Świderek 1
Affiliation
|
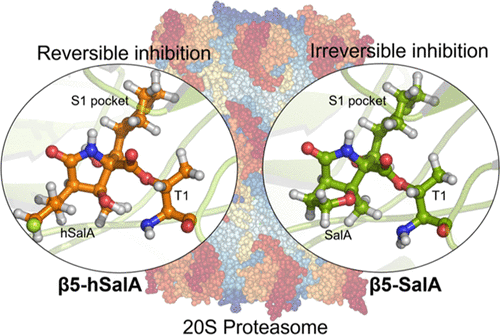
Covalent inhibition of the 20S proteasome is one of the strategies used in the fight against cancer, Chagas’ disease, malaria, tuberculosis, or neurodegenerative disorders. In this work, studies based on quantum mechanics/molecular mechanics (M06-2X/AMBER) molecular dynamics simulations reveal, in agreement with experiments, that homosalinosporamide A (hSalA), contrary to the natural analogue, salinosporamide A (SalA), inhibits the β5 subunit in a reversible manner. The computed free energy landscapes indicate that either SalA or hSalA blocks the active site of the enzyme by a three-step process that includes two common chemical transformations. The last step of the inhibition with SalA, which is an intramolecular cyclization step that defines its irreversible character, was found to be energetically prohibited in the case of hSalA. In contrast, an additional chemical step was found that ensures neutralization of the negative charge accumulated during the β-lactone ring opening, thus providing a stable product of inhibition that agrees with X-ray diffraction studies. The weaker stabilization of this final product explains its reversible character that was further confirmed by exploring possible paths of hydrolysis of the covalent adduct. Finally, the rate-limiting step in both inhibitors corresponds to the nucleophilic attack of Thr1 on C1 carbon of the β-lactone. The identical activation free energies computed for SalA and hSalA agree with the undistinguishable experimentally measured values of IC50.
中文翻译:

Salinosporamide A 和 Homosalinosporamide A 在人 20S 蛋白酶体共价抑制中不同可逆特性的起源
20S 蛋白酶体的共价抑制是用于对抗癌症、南美锥虫病、疟疾、肺结核或神经退行性疾病的策略之一。在这项工作中,基于量子力学/分子力学 (M06-2X/AMBER) 分子动力学模拟的研究表明,与实验一致,高盐孢酰胺 A (hSalA) 与天然类似物盐孢酰胺 A (SalA) 相反,抑制了β5 亚基可逆。计算出的自由能图表明,SalA 或 hSalA 通过包括两个常见化学转化的三步过程来阻断酶的活性位点。SalA 抑制的最后一步,即定义其不可逆特性的分子内环化步骤,在 hSalA 的情况下被发现在能量上被禁止。相比之下,发现了一个额外的化学步骤,可确保中和 β-内酯开环期间积累的负电荷,从而提供与 X 射线衍射研究一致的稳定抑制产物。该最终产物较弱的稳定性解释了其可逆特性,通过探索共价加合物水解的可能途径进一步证实了这一特性。最后,两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致 从而提供与 X 射线衍射研究一致的稳定抑制产物。该最终产物较弱的稳定性解释了其可逆特性,通过探索共价加合物水解的可能途径进一步证实了这一特性。最后,两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致 从而提供与 X 射线衍射研究一致的稳定抑制产物。该最终产物较弱的稳定性解释了其可逆特性,通过探索共价加合物水解的可能途径进一步证实了这一特性。最后,两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致 两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致 两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致50 .
更新日期:2021-09-17
中文翻译:
Salinosporamide A 和 Homosalinosporamide A 在人 20S 蛋白酶体共价抑制中不同可逆特性的起源
20S 蛋白酶体的共价抑制是用于对抗癌症、南美锥虫病、疟疾、肺结核或神经退行性疾病的策略之一。在这项工作中,基于量子力学/分子力学 (M06-2X/AMBER) 分子动力学模拟的研究表明,与实验一致,高盐孢酰胺 A (hSalA) 与天然类似物盐孢酰胺 A (SalA) 相反,抑制了β5 亚基可逆。计算出的自由能图表明,SalA 或 hSalA 通过包括两个常见化学转化的三步过程来阻断酶的活性位点。SalA 抑制的最后一步,即定义其不可逆特性的分子内环化步骤,在 hSalA 的情况下被发现在能量上被禁止。相比之下,发现了一个额外的化学步骤,可确保中和 β-内酯开环期间积累的负电荷,从而提供与 X 射线衍射研究一致的稳定抑制产物。该最终产物较弱的稳定性解释了其可逆特性,通过探索共价加合物水解的可能途径进一步证实了这一特性。最后,两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致 从而提供与 X 射线衍射研究一致的稳定抑制产物。该最终产物较弱的稳定性解释了其可逆特性,通过探索共价加合物水解的可能途径进一步证实了这一特性。最后,两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致 从而提供与 X 射线衍射研究一致的稳定抑制产物。该最终产物较弱的稳定性解释了其可逆特性,通过探索共价加合物水解的可能途径进一步证实了这一特性。最后,两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致 两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致 两种抑制剂中的限速步骤对应于 Thr1 对 β-内酯的 C1 碳的亲核攻击。为 SalA 和 hSalA 计算的相同激活自由能与 IC 无法区分的实验测量值一致50 .

























 京公网安备 11010802027423号
京公网安备 11010802027423号