当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Mechanistic Insights into CO2 Electroreduction on Ni2P: Understanding Its Selectivity toward Multicarbon Products
ACS Catalysis ( IF 12.9 ) Pub Date : 2021-09-07 , DOI: 10.1021/acscatal.1c03639 Sayan Banerjee 1 , Arvin Kakekhani 1 , Robert B. Wexler 2 , Andrew M. Rappe 1
ACS Catalysis ( IF 12.9 ) Pub Date : 2021-09-07 , DOI: 10.1021/acscatal.1c03639 Sayan Banerjee 1 , Arvin Kakekhani 1 , Robert B. Wexler 2 , Andrew M. Rappe 1
Affiliation
|
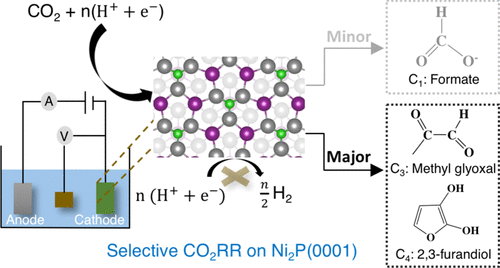
Recently, nickel phosphides (NixPy) have been reported to enable selective electrochemical formation of multicarbon products (C3 and C4) via the CO2 reduction reaction (CO2RR); nevertheless, their activities remain low. In order to understand the roots of their high selectivity and low activity and to direct the design of more active NixPy-based CO2RR catalysts, we investigate the CO2RR mechanism on Ni2P using density functional theory (DFT) calculations. We reveal that the reaction proceeds through the formate pathway, followed by formaldehyde (H2CO*) formation and self-condensation. Moreover, we demonstrate that surface hydride transfer steps, along with surface-mediated C–C coupling, are essential in order to avoid C1 product formation and boost selectivity toward multicarbon products. In addition, we find that the thermal surface hydride transfer from the surface to the physisorbed CO2 is one of the key rate-limiting steps, and since it is not electroactive, it cannot be accelerated by applying an overpotential. Finally, our results also show that the hydrogen affinity of the surface and the dynamic surface reconstruction via H adsorption facilitate selective CO2 reduction and C–C coupling on Ni2P. These findings provide an impetus for exploring materials design space to identify the physical principles that govern the thermodynamics of rate-limiting thermal steps in electrocatalytic processes.
中文翻译:

Ni2P 上 CO2 电还原的机理洞察:了解其对多碳产品的选择性
近来,镍磷化物(镍X P Ý)已经报道,使的多碳制品(C选择性电化学形成3和C 4)通过将CO 2还原反应(CO 2 RR); 尽管如此,他们的活动仍然很少。为了了解其高选择性和低活性的根源并指导设计更具活性的 Ni x P y基 CO 2 RR 催化剂,我们研究了Ni 2上的 CO 2 RR 机理P 使用密度泛函理论 (DFT) 计算。我们发现反应通过甲酸盐途径进行,然后是甲醛 (H 2 CO*) 形成和自缩合。此外,我们证明表面氢化物转移步骤以及表面介导的 C-C 偶联对于避免 C 1产物形成和提高对多碳产物的选择性至关重要。此外,我们发现热表面氢化物从表面转移到物理吸附的 CO 2是关键的限速步骤之一,并且由于它没有电活性,因此不能通过施加过电位来加速。最后,我们的结果还表明,表面的氢亲和力和动态表面重建通过H 吸附促进了 Ni 2 P上的选择性 CO 2还原和 C-C 耦合。这些发现为探索材料设计空间提供了动力,以确定控制电催化过程中限速热步骤的热力学的物理原理。
更新日期:2021-09-17
中文翻译:
Ni2P 上 CO2 电还原的机理洞察:了解其对多碳产品的选择性
近来,镍磷化物(镍X P Ý)已经报道,使的多碳制品(C选择性电化学形成3和C 4)通过将CO 2还原反应(CO 2 RR); 尽管如此,他们的活动仍然很少。为了了解其高选择性和低活性的根源并指导设计更具活性的 Ni x P y基 CO 2 RR 催化剂,我们研究了Ni 2上的 CO 2 RR 机理P 使用密度泛函理论 (DFT) 计算。我们发现反应通过甲酸盐途径进行,然后是甲醛 (H 2 CO*) 形成和自缩合。此外,我们证明表面氢化物转移步骤以及表面介导的 C-C 偶联对于避免 C 1产物形成和提高对多碳产物的选择性至关重要。此外,我们发现热表面氢化物从表面转移到物理吸附的 CO 2是关键的限速步骤之一,并且由于它没有电活性,因此不能通过施加过电位来加速。最后,我们的结果还表明,表面的氢亲和力和动态表面重建通过H 吸附促进了 Ni 2 P上的选择性 CO 2还原和 C-C 耦合。这些发现为探索材料设计空间提供了动力,以确定控制电催化过程中限速热步骤的热力学的物理原理。

























 京公网安备 11010802027423号
京公网安备 11010802027423号