当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Effects of All-Atom Molecular Mechanics Force Fields on Amyloid Peptide Assembly: The Case of PHF6 Peptide of Tau Protein
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-07 , DOI: 10.1021/acs.jctc.1c00028 Viet Hoang Man 1 , Xibing He 1 , Jie Gao 2 , Junmei Wang 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-07 , DOI: 10.1021/acs.jctc.1c00028 Viet Hoang Man 1 , Xibing He 1 , Jie Gao 2 , Junmei Wang 1
Affiliation
|
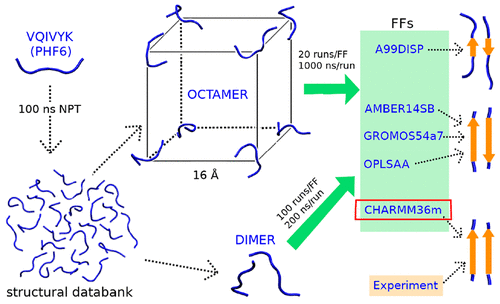
Molecular dynamics (MD) simulations play a vital role in revealing the mechanism of amyloid aggregation that is crucial to the therapeutic agent development for Alzheimer’s Disease. However, the accuracy of MD simulation results strongly depends on the force field employed. In our previous benchmark for 17 all-atom force fields on modeling of amyloid aggregation using the Aβ16–22 dimer, we showed that AMBER14SB and CHARMM36m are suitable force fields for amyloid aggregation simulation, while GROMOS54a7 and OPLSAA are not good for the task. In this work, we continue assessing the applicability of atomistic force fields on amyloid aggregation using the VQIVYK (PHF6) peptide which is essential for tau-protein aggregation. Although, both Aβ16–22 and PHF6 peptides formed fibrils in vitro, the PHF6 fibrils are parallel β-sheets, while the Aβ16–22 fibrils are antiparallel β-sheets. We performed an all-atom large-scale MD simulation in explicit water on the PHF6 dimer and octa-peptides systems using five mainstream force fields, including AMBER99SB-disp, AMBER14SB, CHARMM36m, GROMOS54a7, and OPLSAA. The accumulated simulation time is 0.2 ms. Our result showed that the β-sheet structures of PHF6 peptides sampled by AMBER99SB-disp, AMBER14SB, GROMOS54a7, and OPLSAA are in favor of the antiparallel β-sheets, while the dominant type of β-sheet structures is parallel β-sheet by using CHARMM36m. Among the five force fields, CHARMM36m provides the strongest CH−π interaction that was observed in an NMR study. The comparison between our results and experimental observation indicates that CHARMM36m achieved the best performance on modeling the aggregation of PHF6 peptides. In summary, CHARMM36m is currently the most suitable force field for studying the aggregation of both amyloid-β and Tau through MD simulations.
中文翻译:

全原子分子力学力场对淀粉样肽组装的影响:以 Tau 蛋白的 PHF6 肽为例
分子动力学 (MD) 模拟在揭示对阿尔茨海默病治疗药物开发至关重要的淀粉样蛋白聚集机制方面发挥着至关重要的作用。然而,MD 模拟结果的准确性很大程度上取决于所采用的力场。在我们之前使用 Aβ 16-22二聚体对淀粉样蛋白聚集建模的 17 个全原子力场的基准测试中,我们表明 AMBER14SB 和 CHARMM36m 是淀粉样蛋白聚集模拟的合适力场,而 GROMOS54a7 和 OPLSAA 不适合该任务。在这项工作中,我们继续使用对 tau 蛋白聚集至关重要的 VQIVYK (PHF6) 肽评估原子力场对淀粉样蛋白聚集的适用性。虽然,Aβ 16-22和 PHF6 肽都形成了原纤维在体外,PHF6 原纤维是平行的 β-折叠,而 Aβ 16-22原纤维是反平行的β-折叠。我们使用五个主流力场,包括 AMBER99SB-disp、AMBER14SB、CHARMM36m、GROMOS54a7 和 OPLSAA,在 PHF6 二聚体和八肽系统上进行了全原子大规模 MD 模拟。累计仿真时间为 0.2 ms。我们的结果表明,通过 AMBER99SB-disp、AMBER14SB、GROMOS54a7 和 OPLSAA 采样的 PHF6 肽的 β-折叠结构有利于反平行 β-折叠,而主要类型的 β-折叠结构是平行 β-折叠。 CHARMM36m。在五个力场中,CHARMM36m 提供了在 NMR 研究中观察到的最强 CH-π 相互作用。我们的结果与实验观察结果的比较表明,CHARMM36m 在模拟 PHF6 肽的聚集方面取得了最佳性能。总之,
更新日期:2021-10-12
中文翻译:
全原子分子力学力场对淀粉样肽组装的影响:以 Tau 蛋白的 PHF6 肽为例
分子动力学 (MD) 模拟在揭示对阿尔茨海默病治疗药物开发至关重要的淀粉样蛋白聚集机制方面发挥着至关重要的作用。然而,MD 模拟结果的准确性很大程度上取决于所采用的力场。在我们之前使用 Aβ 16-22二聚体对淀粉样蛋白聚集建模的 17 个全原子力场的基准测试中,我们表明 AMBER14SB 和 CHARMM36m 是淀粉样蛋白聚集模拟的合适力场,而 GROMOS54a7 和 OPLSAA 不适合该任务。在这项工作中,我们继续使用对 tau 蛋白聚集至关重要的 VQIVYK (PHF6) 肽评估原子力场对淀粉样蛋白聚集的适用性。虽然,Aβ 16-22和 PHF6 肽都形成了原纤维在体外,PHF6 原纤维是平行的 β-折叠,而 Aβ 16-22原纤维是反平行的β-折叠。我们使用五个主流力场,包括 AMBER99SB-disp、AMBER14SB、CHARMM36m、GROMOS54a7 和 OPLSAA,在 PHF6 二聚体和八肽系统上进行了全原子大规模 MD 模拟。累计仿真时间为 0.2 ms。我们的结果表明,通过 AMBER99SB-disp、AMBER14SB、GROMOS54a7 和 OPLSAA 采样的 PHF6 肽的 β-折叠结构有利于反平行 β-折叠,而主要类型的 β-折叠结构是平行 β-折叠。 CHARMM36m。在五个力场中,CHARMM36m 提供了在 NMR 研究中观察到的最强 CH-π 相互作用。我们的结果与实验观察结果的比较表明,CHARMM36m 在模拟 PHF6 肽的聚集方面取得了最佳性能。总之,

























 京公网安备 11010802027423号
京公网安备 11010802027423号