当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Machine Learning to Predict Diels–Alder Reaction Barriers from the Reactant State Electron Density
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-03 , DOI: 10.1021/acs.jctc.1c00623 Santiago Vargas 1 , Matthew R Hennefarth 1 , Zhihao Liu 1 , Anastassia N Alexandrova 1, 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-03 , DOI: 10.1021/acs.jctc.1c00623 Santiago Vargas 1 , Matthew R Hennefarth 1 , Zhihao Liu 1 , Anastassia N Alexandrova 1, 2
Affiliation
|
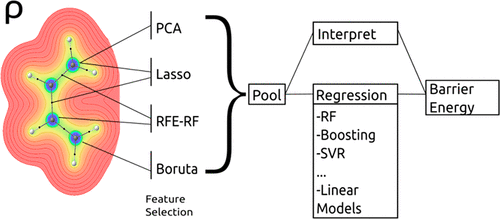
Reaction barriers are key to our understanding of chemical reactivity and catalysis. Certain reactions are so seminal in chemistry that countless variants, with or without catalysts, have been studied, and their barriers have been computed or measured experimentally. This wealth of data represents a perfect opportunity to leverage machine learning models, which could quickly predict barriers without explicit calculations or measurement. Here, we show that the topological descriptors of the quantum mechanical charge density in the reactant state constitute a set that is both rigorous and continuous and can be used effectively for the prediction of reaction barrier energies to a high degree of accuracy. We demonstrate this on the Diels–Alder reaction, highly important in biology and medicinal chemistry, and as such, studied extensively. This reaction exhibits a range of barriers as large as 270 kJ/mol. While we trained our single-objective supervised (labeled) regression algorithms on simpler Diels–Alder reactions in solution, they predict reaction barriers also in significantly more complicated contexts, such a Diels–Alder reaction catalyzed by an artificial enzyme and its evolved variants, in agreement with experimental changes in kcat. We expect this tool to apply broadly to a variety of reactions in solution or in the presence of a catalyst, for screening and circumventing heavily involved computations or experiments.
中文翻译:

机器学习从反应物状态电子密度预测 Diels-Alder 反应势垒
反应屏障是我们理解化学反应性和催化作用的关键。某些反应在化学中非常具有开创性,以至于研究了无数有或没有催化剂的变体,并且已经通过实验计算或测量了它们的障碍。如此丰富的数据代表了利用机器学习模型的绝佳机会,该模型无需明确计算或测量即可快速预测障碍。在这里,我们展示了反应物状态下量子力学电荷密度的拓扑描述符构成了一个既严格又连续的集合,可以有效地用于高度准确地预测反应势垒能量。我们在 Diels-Alder 反应中证明了这一点,这在生物学和药物化学中非常重要,因此被广泛研究。该反应表现出高达 270 kJ/mol 的一系列势垒。虽然我们在溶液中更简单的 Diels-Alder 反应上训练了我们的单目标监督(标记)回归算法,但它们也预测了在更复杂的环境中的反应障碍,例如由人工酶及其进化变体催化的 Diels-Alder 反应,在与实验变化一致ķ猫。我们希望该工具能够广泛应用于溶液中或催化剂存在下的各种反应,以筛选和规避大量涉及的计算或实验。
更新日期:2021-10-12
中文翻译:
机器学习从反应物状态电子密度预测 Diels-Alder 反应势垒
反应屏障是我们理解化学反应性和催化作用的关键。某些反应在化学中非常具有开创性,以至于研究了无数有或没有催化剂的变体,并且已经通过实验计算或测量了它们的障碍。如此丰富的数据代表了利用机器学习模型的绝佳机会,该模型无需明确计算或测量即可快速预测障碍。在这里,我们展示了反应物状态下量子力学电荷密度的拓扑描述符构成了一个既严格又连续的集合,可以有效地用于高度准确地预测反应势垒能量。我们在 Diels-Alder 反应中证明了这一点,这在生物学和药物化学中非常重要,因此被广泛研究。该反应表现出高达 270 kJ/mol 的一系列势垒。虽然我们在溶液中更简单的 Diels-Alder 反应上训练了我们的单目标监督(标记)回归算法,但它们也预测了在更复杂的环境中的反应障碍,例如由人工酶及其进化变体催化的 Diels-Alder 反应,在与实验变化一致ķ猫。我们希望该工具能够广泛应用于溶液中或催化剂存在下的各种反应,以筛选和规避大量涉及的计算或实验。
























 京公网安备 11010802027423号
京公网安备 11010802027423号