当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
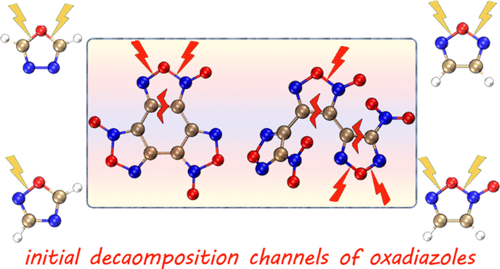
Initial Steps and Thermochemistry of Unimolecular Decomposition of Oxadiazole Energetic Materials: Quantum Chemistry Modeling
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2021-09-01 , DOI: 10.1021/acs.jpca.1c05876 Shuangfei Zhu 1 , Wei Yang 2 , Qiang Gan 1 , Changgen Feng 1
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2021-09-01 , DOI: 10.1021/acs.jpca.1c05876 Shuangfei Zhu 1 , Wei Yang 2 , Qiang Gan 1 , Changgen Feng 1
Affiliation
|
In order to resolve the existing discrepancies in the mechanism and key intermediates of oxadiazole thermolysis, the initial decomposition pathways of oxadiazoles have been studied comprehensively using the M062X method for optimization and CBS-QB3 and DLPNO-CCSD(T) methods for energies. The transformation from the furoxan ring to nitro group was suggested as a potential decay channel of furoxan compounds. Results of thermochemistry calculations showed that the preferred decomposition reaction of oxadiazoles is the ring-opening through the cleavage of the O–C or O–N bond. The introduction of the nitro group has little effect on the preferential path of oxadiazole thermal decomposition, but a great impact on the energy barrier. The lowest energy barrier and bond dissociation energy of NO2 loss of azoles were comprehensively studied based on the quantum chemistry calculations. The initial decay steps of 3,4-dinitrofurazanfuroxan and benzotrifuroxan were also studied to give insights into the mechanism of primary stages of thermal decomposition of oxadiazoles.
中文翻译:

恶二唑高能材料单分子分解的初始步骤和热化学:量子化学建模
为解决恶二唑热解机理和关键中间体存在的差异,利用M062X优化方法、CBS-QB3和DLPNO-CCSD(T)方法对恶二唑的初始分解途径进行了综合研究。从呋喃环到硝基的转变被认为是呋喃化合物的潜在衰变通道。热化学计算结果表明,恶二唑的首选分解反应是通过 O-C 或 O-N 键的断裂开环。硝基的引入对恶二唑热分解的优先路径影响不大,但对能垒影响很大。NO 2的最低能垒和键解离能基于量子化学计算,综合研究了唑类的损失。还研究了 3,4-二硝基呋喃并呋喃和苯并三呋喃的初始衰变步骤,以深入了解恶二唑热分解初级阶段的机制。
更新日期:2021-09-16
中文翻译:
恶二唑高能材料单分子分解的初始步骤和热化学:量子化学建模
为解决恶二唑热解机理和关键中间体存在的差异,利用M062X优化方法、CBS-QB3和DLPNO-CCSD(T)方法对恶二唑的初始分解途径进行了综合研究。从呋喃环到硝基的转变被认为是呋喃化合物的潜在衰变通道。热化学计算结果表明,恶二唑的首选分解反应是通过 O-C 或 O-N 键的断裂开环。硝基的引入对恶二唑热分解的优先路径影响不大,但对能垒影响很大。NO 2的最低能垒和键解离能基于量子化学计算,综合研究了唑类的损失。还研究了 3,4-二硝基呋喃并呋喃和苯并三呋喃的初始衰变步骤,以深入了解恶二唑热分解初级阶段的机制。
























 京公网安备 11010802027423号
京公网安备 11010802027423号