Topics in Catalysis ( IF 3.6 ) Pub Date : 2021-08-25 , DOI: 10.1007/s11244-021-01501-5 Li-Juan Yu 1 , Mitchell T. Blyth 1 , Michelle L. Coote 1
|
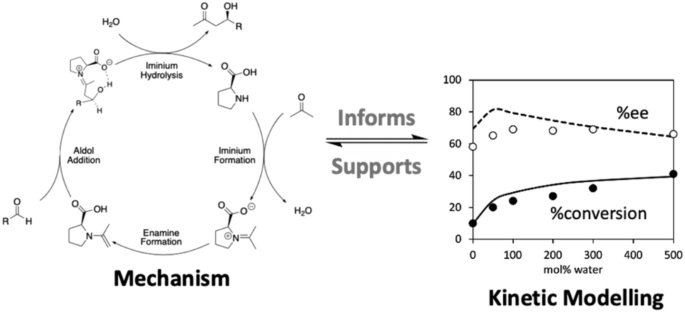
The full catalytic cycle of the proline-catalyzed intermolecular aldol reaction of acetone and p-nitrobenzaldehyde in acetone solvent has been investigated by quantum chemistry at the G3(MP2,CC)//M062X/6–31+G(d)/SMD level of theory, and the results used to develop an ab initio kinetic model. Proline catalyzes the aldol reaction according to the enamine mechanism. The initial reaction between proline and acetone was reinvestigated, and a revised mechanism for enamine formation is proposed in which a second proline assists the process contributing to the enamine formation. Using various initial concentrations of proline while keeping the experimental ratio of water, aldehyde and acetone constant, we find that the enamine formation from the first-order to proline pathway dominates when the concentration of proline is low (< 0.005 M); while the second-order enamine formation pathways contribute and then dominate as the proline concentration is increased. The relative rates of formation of the syn and anti-enamine are not important, as these interconvert via C–N bond rotation and equilibrate faster than their subsequent reaction, which follows the standard Houk/List mechanism. While the stereochemistry can be predicted from an analysis of the alternative C–C bond formation pathways, their relative contributions to the major and minor product yields are influenced by their subsequent rates of hydrolysis. Indeed, while C–C bond formation is normally considered rate determining, our kinetic simulations show that the kinetic model is more complicated than this and under typically used concentrations, the process of initial enamine formation, C–C bond formation and the initial stages of product release all contribute to the overall reaction rate. Using our kinetic model, we predict that yield and %ee are optimal for concentrations of [proline] = 0.005 M, [acetone] = 2.25 M, [aldehyde] = 0.1 M, and [water] = 0.6 M. Using excess acetone (up to 2.6 M) increases both conversion and %ee. Excess aldehyde increases %ee but decreases conversion, and excess catalyst increases the conversion but decreases %ee. Aside from the indirect effect of increasing the solubility of the proline catalyst, water increases both conversion and %ee up to a point, but at large concentrations (> 1.0 M) excess water is expected to decrease %ee. Side reactivity, including aldol condensation, acetone self-aldolization, oxazolidinone formation and azomethine and 1-oxapyrrolizidine formation were all considered in our kinetic model but shown to have a negligible effect (< 2%) on the yield and %ee over the full range of reaction conditions investigated.
Graphic Abstract
中文翻译:
脯氨酸催化的分子间羟醛反应的重新研究:从头算动力学建模研究
通过量子化学在 G3(MP2,CC)//M062X/6–31+G(d)/SMD 水平上研究了脯氨酸催化丙酮和对硝基苯甲醛在丙酮溶剂中的分子间羟醛反应的完整催化循环理论,以及用于开发 ab initio 动力学模型的结果。脯氨酸根据烯胺机制催化羟醛反应。重新研究了脯氨酸和丙酮之间的初始反应,并提出了一种修正的烯胺形成机制,其中第二个脯氨酸有助于促进烯胺形成的过程。使用各种初始浓度的脯氨酸,同时保持水、醛和丙酮的实验比例恒定,我们发现当脯氨酸浓度较低 (< 0.005 M) 时,从一级到脯氨酸途径的烯胺形成占主导地位;而随着脯氨酸浓度的增加,二级烯胺形成途径有所贡献,然后占主导地位。相对形成率SYN和反-enamine 并不重要,因为它们通过 C-N 键旋转相互转化,并且比随后的反应更快地平衡,这遵循标准的 Houk/List 机制。虽然立体化学可以通过对替代 C-C 键形成途径的分析来预测,但它们对主要和次要产物产率的相对贡献受其随后水解速率的影响。事实上,虽然 C-C 键的形成通常被认为是速率决定因素,但我们的动力学模拟表明,动力学模型比这更复杂,并且在通常使用的浓度下,初始烯胺形成过程、C-C 键形成和初始阶段产品的释放都会影响整体反应速度。使用我们的动力学模型,我们预测产率和 %ee 对于 [脯氨酸] = 0.005 M 的浓度是最佳的,[丙酮] = 2.25 M,[醛] = 0.1 M,[水] = 0.6 M。使用过量丙酮(最多 2.6 M)可提高转化率和 %ee。过量的醛会增加 %ee 但会降低转化率,过量的催化剂会增加转化率但会降低 %ee。除了增加脯氨酸催化剂溶解度的间接影响外,水在一定程度上提高了转化率和 %ee,但在高浓度 (> 1.0 M) 下,预计过量的水会降低 %ee。副反应性,包括醛醇缩合、丙酮自醛缩、恶唑烷酮形成和偶氮甲碱和 1-氧杂吡咯里西啶形成都在我们的动力学模型中被考虑在内,但在整个范围内对产率和 %ee 的影响可以忽略不计(< 2%)研究的反应条件。使用过量的丙酮(最多 2.6 M)可提高转化率和 %ee。过量的醛会增加 %ee 但会降低转化率,过量的催化剂会增加转化率但会降低 %ee。除了增加脯氨酸催化剂溶解度的间接影响外,水在一定程度上提高了转化率和 %ee,但在高浓度 (> 1.0 M) 下,预计过量的水会降低 %ee。副反应性,包括醛醇缩合、丙酮自醛缩、恶唑烷酮形成和偶氮甲碱和 1-氧杂吡咯里西啶形成都在我们的动力学模型中被考虑在内,但在整个范围内对产率和 %ee 的影响可以忽略不计(< 2%)研究的反应条件。使用过量的丙酮(最多 2.6 M)可提高转化率和 %ee。过量的醛会增加 %ee 但会降低转化率,过量的催化剂会增加转化率但会降低 %ee。除了增加脯氨酸催化剂溶解度的间接影响外,水在一定程度上提高了转化率和 %ee,但在高浓度 (> 1.0 M) 下,预计过量的水会降低 %ee。副反应性,包括醛醇缩合、丙酮自醛缩、恶唑烷酮形成和偶氮甲碱和 1-氧杂吡咯里西啶形成都在我们的动力学模型中被考虑在内,但在整个范围内对产率和 %ee 的影响可以忽略不计(< 2%)研究的反应条件。过量的催化剂会增加转化率但会降低 %ee。除了增加脯氨酸催化剂溶解度的间接影响外,水在一定程度上提高了转化率和 %ee,但在高浓度 (> 1.0 M) 下,预计过量的水会降低 %ee。副反应性,包括醛醇缩合、丙酮自醛缩、恶唑烷酮形成和偶氮甲碱和 1-氧杂吡咯里西啶形成都在我们的动力学模型中被考虑在内,但在整个范围内对产率和 %ee 的影响可以忽略不计(< 2%)研究的反应条件。过量的催化剂会提高转化率,但会降低 %ee。除了增加脯氨酸催化剂溶解度的间接影响外,水在一定程度上提高了转化率和 %ee,但在高浓度 (> 1.0 M) 下,预计过量的水会降低 %ee。副反应性,包括醛醇缩合、丙酮自醛缩、恶唑烷酮形成和偶氮甲碱和 1-氧杂吡咯里西啶形成都在我们的动力学模型中被考虑在内,但在整个范围内对产率和 %ee 的影响可以忽略不计(< 2%)研究的反应条件。0 M) 过量的水预计会降低 %ee。副反应性,包括醛醇缩合、丙酮自醛缩、恶唑烷酮形成和偶氮甲碱和 1-氧杂吡咯里西啶形成都在我们的动力学模型中被考虑在内,但在整个范围内对产率和 %ee 的影响可以忽略不计(< 2%)研究的反应条件。0 M) 过量的水预计会降低 %ee。副反应性,包括醛醇缩合、丙酮自醛缩、恶唑烷酮形成和偶氮甲碱和 1-氧杂吡咯里西啶形成都在我们的动力学模型中被考虑在内,但在整个范围内对产率和 %ee 的影响可以忽略不计(< 2%)研究的反应条件。

























 京公网安备 11010802027423号
京公网安备 11010802027423号