Surface Science ( IF 1.9 ) Pub Date : 2021-08-20 , DOI: 10.1016/j.susc.2021.121919 Wenjie Yang 1 , Zihan Yan 1 , Kangjin Zhang 1 , Wenyan Wang 1 , Shuqi Lei 1 , Shuming Zeng 1 , Yusong Tu 1, 2
|
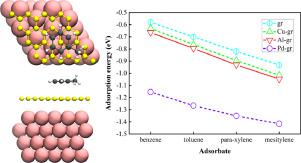
We perform a comparative theoretical study of benzene and its homologues (toluene, para-xylene, and mesitylene) adsorbed on Cu/Al/Pd(111)-supported graphene using density functional theory (DFT). The long-range attraction is handled by semiempirical dispersion correction method (PBE-D3) and ab initio van der Waals density functionals (vdW-DF2, optB86b-vdW, optB88-vdW, and SCAN-rVV10). Our results show a systematic increase in the adsorption energy of organic molecules on graphene in the presence of underlaying metal substrates. In the case of strong metal-graphene contact, such as Pd(111)-graphene, the adsorption energy of benzene even increases 0.44 eV, which presents suitable platform for filtering harmful molecules such as benzene. Besides, we find out that the dispersion-corrected functionals play a fundamental role in the structure and adsorption energy. Furthermore, the adsorption energy increases linearly with the number of methyl groups on the benzene ring.
中文翻译:
石墨烯-金属表面苯及其同系物的第一性原理研究:伦敦色散校正比较
我们使用密度泛函理论 (DFT) 对吸附在 Cu/Al/Pd(111) 支撑的石墨烯上的苯及其同系物(甲苯、对二甲苯和均三甲苯)进行比较理论研究。通过半经验色散校正方法 (PBE-D3) 和ab initio处理远程吸引力范德华密度泛函(vdW-DF2、optB86b-vdW、optB88-vdW 和 SCAN-rVV10)。我们的结果表明,在底层金属基板存在的情况下,有机分子在石墨烯上的吸附能系统地增加。在金属-石墨烯强接触的情况下,如Pd(111)-石墨烯,苯的吸附能甚至增加0.44 eV,为过滤苯等有害分子提供了合适的平台。此外,我们发现色散校正泛函在结构和吸附能中起着重要作用。此外,吸附能随着苯环上甲基的数量线性增加。

























 京公网安备 11010802027423号
京公网安备 11010802027423号