当前位置:
X-MOL 学术
›
Energy Fuels
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Impact of Asphaltenes on the Adsorption Behavior of Petroleum Vanadyl Porphyrins: Kinetic and Thermodynamic Aspects
Energy & Fuels ( IF 5.3 ) Pub Date : 2021-08-16 , DOI: 10.1021/acs.energyfuels.1c01495 Nikolay Mironov 1 , Dmitry Milordov 1 , Elvira Tazeeva 1 , Damir Tazeev 1 , Guzalia Abilova 1 , Svetlana Yakubova 1 , Makhmut Yakubov 1
Energy & Fuels ( IF 5.3 ) Pub Date : 2021-08-16 , DOI: 10.1021/acs.energyfuels.1c01495 Nikolay Mironov 1 , Dmitry Milordov 1 , Elvira Tazeeva 1 , Damir Tazeev 1 , Guzalia Abilova 1 , Svetlana Yakubova 1 , Makhmut Yakubov 1
Affiliation
|
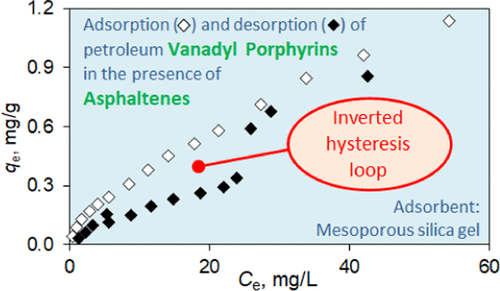
In the present work, the first data on the adsorption behavior of petroleum vanadyl porphyrins in the presence and absence of asphaltenes are afforded. As adsorptives, N,N′-dimethylformamide extract of asphaltenes containing 9.88 wt % vanadyl porphyrins and high-purity vanadyl porphyrins preisolated from the extract by the sulfocationite-based chromatographic method were used. As adsorbents, two mesoporous silica gels distinguished in their ability to slow down the rate of diffusion of asphaltene nanoaggregates were chosen. Changes in adsorptive concentration upon adsorbing were monitored spectrophotometrically. Time- and concentration-dependent adsorption studies have been conducted, and particular features in the adsorption behavior of asphaltenes and vanadyl porphyrins have been revealed and used in modeling the intermolecular asphaltene–vanadyl porphyrin interactions occurring inside the adsorbent pores. An adsorption kinetic experiment showed that the asphaltenes diffuse inside the adsorbent pores in the aggregated form and reduce the diffusion rate of vanadyl porphyrins virtually up to the rate of the asphaltene nanoaggregates themselves. An equilibrium adsorption experiment revealed that when a Langmuir-type adsorption happens the asphaltene nanoaggregates adsorbed are composed of 5–8 monomers, which is well consistent with the Yen–Mullins model. No competition for free adsorption sites between asphaltenes and vanadyl porphyrins was observed, which indicates a coaggregation mechanism of vanadyl porphyrin uptake. This assumption was supported by an adsorption thermodynamic study displaying that enthalpy change of adsorption obtained by the van’t Hoff method takes positive values for both the asphaltenes and vanadyl porphyrins present in them (ΔH° > 0) but not for isolated vanadyl porphyrins (ΔH° < 0). An average molecular weight of asphaltene monomers required for thermodynamic calculations was derived from their matrix-assisted laser desorption/ionization (MALDI) mass spectrum mathematically simulated using a log–normal distribution function. The results of the present work contribute to better understanding the nature of asphaltene–petroporphyrin interactions responsible for aggregation and adsorption properties of these petroleum components.
中文翻译:

沥青质对石油氧钒卟啉吸附行为的影响:动力学和热力学方面
在目前的工作中,提供了在存在和不存在沥青质的情况下石油氧钒卟啉的吸附行为的第一个数据。作为吸附剂,N , N使用含有 9.88 wt% 氧钒卟啉的 '-二甲基甲酰胺提取物和通过基于磺基阳离子的色谱法从提取物中预分离的高纯度氧钒卟啉。作为吸附剂,选择了两种在减缓沥青质纳米聚集体扩散速度方面表现突出的中孔硅胶。通过分光光度法监测吸附后吸附浓度的变化。已经进行了与时间和浓度相关的吸附研究,揭示了沥青质和氧钒卟啉吸附行为的特殊特征,并将其用于模拟吸附剂孔隙内发生的分子间沥青质 - 氧钒卟啉相互作用。吸附动力学实验表明,沥青质以聚集形式在吸附剂孔隙内扩散,降低了氧钒卟啉的扩散速率,几乎达到沥青质纳米聚集体本身的速率。平衡吸附实验表明,当发生朗缪尔型吸附时,吸附的沥青质纳米团聚体由 5-8 个单体组成,这与 Yen-Mullins 模型非常一致。没有观察到沥青质和氧钒卟啉之间对自由吸附位点的竞争,这表明氧钒卟啉吸收的共聚机制。这一假设得到了吸附热力学研究的支持,该研究表明,通过 van't Hoff 方法获得的吸附焓变对于其中存在的沥青质和氧钒卟啉均取正值(ΔH ° > 0) 但不适用于分离的氧钒卟啉 (Δ H ° < 0)。热力学计算所需的沥青质单体的平均分子量来自它们的基质辅助激光解吸/电离 (MALDI) 质谱,使用对数正态分布函数进行数学模拟。目前的工作结果有助于更好地理解沥青质-石油卟啉相互作用的性质,这些相互作用负责这些石油组分的聚集和吸附特性。
更新日期:2021-09-16
中文翻译:
沥青质对石油氧钒卟啉吸附行为的影响:动力学和热力学方面
在目前的工作中,提供了在存在和不存在沥青质的情况下石油氧钒卟啉的吸附行为的第一个数据。作为吸附剂,N , N使用含有 9.88 wt% 氧钒卟啉的 '-二甲基甲酰胺提取物和通过基于磺基阳离子的色谱法从提取物中预分离的高纯度氧钒卟啉。作为吸附剂,选择了两种在减缓沥青质纳米聚集体扩散速度方面表现突出的中孔硅胶。通过分光光度法监测吸附后吸附浓度的变化。已经进行了与时间和浓度相关的吸附研究,揭示了沥青质和氧钒卟啉吸附行为的特殊特征,并将其用于模拟吸附剂孔隙内发生的分子间沥青质 - 氧钒卟啉相互作用。吸附动力学实验表明,沥青质以聚集形式在吸附剂孔隙内扩散,降低了氧钒卟啉的扩散速率,几乎达到沥青质纳米聚集体本身的速率。平衡吸附实验表明,当发生朗缪尔型吸附时,吸附的沥青质纳米团聚体由 5-8 个单体组成,这与 Yen-Mullins 模型非常一致。没有观察到沥青质和氧钒卟啉之间对自由吸附位点的竞争,这表明氧钒卟啉吸收的共聚机制。这一假设得到了吸附热力学研究的支持,该研究表明,通过 van't Hoff 方法获得的吸附焓变对于其中存在的沥青质和氧钒卟啉均取正值(ΔH ° > 0) 但不适用于分离的氧钒卟啉 (Δ H ° < 0)。热力学计算所需的沥青质单体的平均分子量来自它们的基质辅助激光解吸/电离 (MALDI) 质谱,使用对数正态分布函数进行数学模拟。目前的工作结果有助于更好地理解沥青质-石油卟啉相互作用的性质,这些相互作用负责这些石油组分的聚集和吸附特性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号