当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Implicit solvation in domain based pair natural orbital coupled cluster (DLPNO-CCSD) theory
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2021-08-04 , DOI: 10.1002/jcc.26726 Miquel Garcia-Ratés 1 , Ute Becker 1 , Frank Neese 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2021-08-04 , DOI: 10.1002/jcc.26726 Miquel Garcia-Ratés 1 , Ute Becker 1 , Frank Neese 1
Affiliation
|
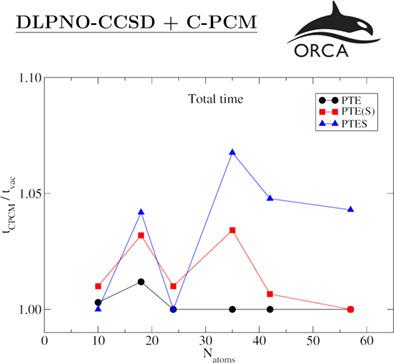
A nearly linear scaling implementation of coupled-cluster with singles and doubles excitations (CCSD) can be achieved by means of the domain-based local pair natural orbital (DLPNO) method. The combination of DLPNO-CCSD with implicit solvation methods allows the calculation of accurate energies and chemical properties of solvated systems at an affordable computational cost. We have efficiently implemented different schemes within the conductor-like polarizable continuum model (C-PCM) for DLPNO-CCSD in the ORCA quantum chemistry suite. In our implementation, the overhead due to the additional solvent terms amounts to less than 5% of the time the equivalent gas phase job takes. Our results for organic neutrals and open-shell ions in water show that for most systems, adding solvation terms to the coupled-cluster amplitudes equations and to the energy leads to small changes in the total energy compared to only considering solvated orbitals and corrections to the reference energy. However, when the solute contains certain functional groups, such as carbonyl or nitrile groups, the changes in the energy are larger and estimated to be around 0.04 and 0.02 kcal/mol for each carbonyl and nitrile group in the solute, respectively. For solutes containing metals, the use of accurate CC/C-PCM schemes is crucial to account for correlation solvation effects. Simultaneously, we have calculated the electrostatic component of the solvation energy for neutrals and ions in water for the different DLPNO-CCSD/C-PCM schemes. We observe negligible changes in the deviation between DLPNO-CCSD and canonical-CCSD data. Here, DLPNO-CCSD results outperform those for Hartree-Fock and density functional theory calculations.
中文翻译:

基于域的对自然轨道耦合簇 (DLPNO-CCSD) 理论中的隐式溶剂化
通过基于域的局部对自然轨道 (DLPNO) 方法,可以实现具有单倍和双倍激发 (CCSD) 的耦合簇的近线性缩放实现。DLPNO-CCSD 与隐式溶剂化方法的组合允许以可承受的计算成本计算溶剂化系统的准确能量和化学性质。我们已经在 ORCA 量子化学套件中的 DLPNO-CCSD 的类导体极化连续体模型 (C-PCM) 中有效地实施了不同的方案。在我们的实施中,由于额外的溶剂项导致的开销不到等效气相作业所需时间的 5%。我们对水中有机中性离子和开壳离子的结果表明,对于大多数系统,与仅考虑溶剂化轨道和参考能量修正相比,将溶剂化项添加到耦合簇振幅方程和能量中会导致总能量的微小变化。然而,当溶质含有某些官能团,如羰基或腈基时,能量的变化更大,估计溶质中每个羰基和腈基的能量变化分别约为 0.04 和 0.02 kcal/mol。对于含有金属的溶质,使用准确的 CC/C-PCM 方案对于解释相关溶剂化效应至关重要。同时,我们计算了不同 DLPNO-CCSD/C-PCM 方案的水中中性和离子的溶剂化能的静电分量。我们观察到 DLPNO-CCSD 和规范 CCSD 数据之间偏差的变化可以忽略不计。这里,
更新日期:2021-08-27
中文翻译:
基于域的对自然轨道耦合簇 (DLPNO-CCSD) 理论中的隐式溶剂化
通过基于域的局部对自然轨道 (DLPNO) 方法,可以实现具有单倍和双倍激发 (CCSD) 的耦合簇的近线性缩放实现。DLPNO-CCSD 与隐式溶剂化方法的组合允许以可承受的计算成本计算溶剂化系统的准确能量和化学性质。我们已经在 ORCA 量子化学套件中的 DLPNO-CCSD 的类导体极化连续体模型 (C-PCM) 中有效地实施了不同的方案。在我们的实施中,由于额外的溶剂项导致的开销不到等效气相作业所需时间的 5%。我们对水中有机中性离子和开壳离子的结果表明,对于大多数系统,与仅考虑溶剂化轨道和参考能量修正相比,将溶剂化项添加到耦合簇振幅方程和能量中会导致总能量的微小变化。然而,当溶质含有某些官能团,如羰基或腈基时,能量的变化更大,估计溶质中每个羰基和腈基的能量变化分别约为 0.04 和 0.02 kcal/mol。对于含有金属的溶质,使用准确的 CC/C-PCM 方案对于解释相关溶剂化效应至关重要。同时,我们计算了不同 DLPNO-CCSD/C-PCM 方案的水中中性和离子的溶剂化能的静电分量。我们观察到 DLPNO-CCSD 和规范 CCSD 数据之间偏差的变化可以忽略不计。这里,


























 京公网安备 11010802027423号
京公网安备 11010802027423号