Materials Today Energy ( IF 9.3 ) Pub Date : 2021-08-02 , DOI: 10.1016/j.mtener.2021.100820 Janpreet Singh 1 , Harpreet Kaur 1 , Gurinder Singh 2 , Surya Kant Tripathi 3
|
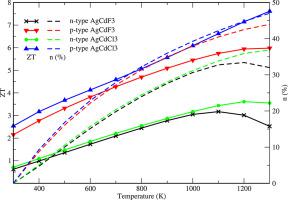
The need for high-performance thermoelectric materials is a hot topic under research to harvest the waste heat generated in the environment. In the present work, we have studied the inorganic halide perovskite AgCdX3 (X = F and Cl) in the cubic phase with the Pm − 3 m space group. We have explored the structural, electronic, elastic, and thermoelectric properties for both materials using first -principles calculations based on density functional theory. The thermoelectric transport coefficients have been calculated from band structure results with the aid of the BoltzTraP2 code based on the semiclassical Boltzmann theory under the constant relaxation time approximation. The deformation potential theory has been implemented to estimate the relaxation time for charge carriers. The Slack equation is solved to evaluate the contribution of lattice thermal conductivity. The results show that the AgCdCl3 has a high value of the figure of merit (ZT) in comparison to that of AgCdF3 at any temperature irrespective of the type of charge carriers. Our findings will provide a guide for the experimentalists to develop high-efficient thermoelectric materials.
中文翻译:
来自第一性原理的立方无机卤化物钙钛矿材料 AgCdX3(X = F 和 Cl)的高热电性能
对高性能热电材料的需求是收集环境中产生的废热的研究热点。在目前的工作中,我们研究了无机卤化物钙钛矿 AgCdX 3(X = F 和 Cl) 在具有 Pm - 3 m 空间群的立方相中。我们使用基于密度泛函理论的第一性原理计算探索了两种材料的结构、电子、弹性和热电特性。在恒定弛豫时间近似下,借助基于半经典玻尔兹曼理论的 BoltzTraP2 代码,根据能带结构结果计算了热电传输系数。变形势理论已被用于估计电荷载流子的弛豫时间。求解松弛方程以评估晶格热导率的贡献。该结果表明,该AgCdCl 3相较于AgCdF具有优值(ZT)的该图的高值3在任何温度下,与电荷载流子的类型无关。我们的发现将为实验人员开发高效热电材料提供指导。

























 京公网安备 11010802027423号
京公网安备 11010802027423号