当前位置:
X-MOL 学术
›
Cryst. Growth Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Assessment of Computational Methods for Calculating Accurate Non-covalent Interaction Energies in 1,2,3,5-Dithiadiazolyl Radicals
Crystal Growth & Design ( IF 3.8 ) Pub Date : 2021-07-30 , DOI: 10.1021/acs.cgd.1c00244 Sahar Nikoo 1 , Jeremy M. Rawson 1
Crystal Growth & Design ( IF 3.8 ) Pub Date : 2021-07-30 , DOI: 10.1021/acs.cgd.1c00244 Sahar Nikoo 1 , Jeremy M. Rawson 1
Affiliation
|
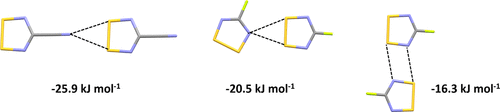
The strength of non-covalent intermolecular interactions between dithiadiazolyl (DTDA) radicals XCNSSN (X = F, Cl, Br, CN) were benchmarked using a high-level post-Hartree–Fock ab initio CCSD(T) theory with a complete basis set and corrected with a counterpoise (CP) correction for basis-set superposition error (BSSE). A range of density functional theory (DFT) functionals and basis sets were then screened to identify appropriate DFT methods which would reflect the benchmark data. These identified that minimal basis sets tended to overestimate the interaction energies, whereas inclusion of additional diffuse and polarization functions led to underestimation of these energies. The application of a CP correction for BSSE generally led to inferior performances in relation to uncorrected data for DFT calculations. The relative strengths of the intermolecular interactions were implemented to rationalize the trends in packing within the series XCNSSN (X = F, Cl, Br, CN). The nature of these interactions was also probed through an Atoms in Molecules approach which reveals intermolecular bond critical points for the different types of interactions, which are all diagnostic of non-covalent interactions between DTDA radicals. These are compared with previous experimental charge-density studies on related DTDA radicals. The best-performing DFT methods were then applied to the α- and β-polymorphs of the larger radical p-NCC6F4CNSSN which was too large for benchmark studies. These confirmed the small energetic differences between α and β phases with the top five best-performing methods and functionals correctly reflecting the relative phase stabilities.
中文翻译:

用于计算 1,2,3,5-二噻二唑基自由基中准确非共价相互作用能的计算方法评估
二噻二唑基 (DTDA) 自由基 XCNSSN (X = F, Cl, Br, CN) 之间非共价分子间相互作用的强度使用具有完整基组的高级后 Hartree-Fock ab initio CCSD(T) 理论进行基准测试并通过对基组叠加误差 (BSSE) 的地网 (CP) 校正进行校正。然后对一系列密度泛函理论 (DFT) 泛函和基组进行筛选,以确定能够反映基准数据的适当 DFT 方法。这些发现最小基组倾向于高估相互作用的能量,而包含额外的漫反射和极化函数会导致对这些能量的低估。对 BSSE 应用 CP 校正通常会导致与 DFT 计算的未校正数据相关的性能较差。实施了分子间相互作用的相对强度,以合理化 XCNSSN (X = F, Cl, Br, CN) 系列内的堆积趋势。这些相互作用的性质也通过分子中的原子方法进行了探索,该方法揭示了不同类型相互作用的分子间键合临界点,这些都是对 DTDA 自由基之间非共价相互作用的诊断。这些与之前对相关 DTDA 自由基的实验电荷密度研究进行了比较。然后将性能最佳的 DFT 方法应用于较大自由基的 α- 和 β-多晶型物 这些相互作用的性质也通过分子中的原子方法进行了探索,该方法揭示了不同类型相互作用的分子间键合临界点,这些都是对 DTDA 自由基之间非共价相互作用的诊断。这些与之前对相关 DTDA 自由基的实验电荷密度研究进行了比较。然后将性能最佳的 DFT 方法应用于较大自由基的 α- 和 β-多晶型物 这些相互作用的性质也通过分子中的原子方法进行了探索,该方法揭示了不同类型相互作用的分子间键合临界点,这些都是对 DTDA 自由基之间非共价相互作用的诊断。这些与之前对相关 DTDA 自由基的实验电荷密度研究进行了比较。然后将性能最佳的 DFT 方法应用于较大自由基的 α- 和 β-多晶型物p -NCC 6 F 4 CNSSN 对于基准研究来说太大了。这些证实了 α 和 β 相之间的微小能量差异,前五种表现最佳的方法和函数正确地反映了相对相稳定性。
更新日期:2021-09-01
中文翻译:
用于计算 1,2,3,5-二噻二唑基自由基中准确非共价相互作用能的计算方法评估
二噻二唑基 (DTDA) 自由基 XCNSSN (X = F, Cl, Br, CN) 之间非共价分子间相互作用的强度使用具有完整基组的高级后 Hartree-Fock ab initio CCSD(T) 理论进行基准测试并通过对基组叠加误差 (BSSE) 的地网 (CP) 校正进行校正。然后对一系列密度泛函理论 (DFT) 泛函和基组进行筛选,以确定能够反映基准数据的适当 DFT 方法。这些发现最小基组倾向于高估相互作用的能量,而包含额外的漫反射和极化函数会导致对这些能量的低估。对 BSSE 应用 CP 校正通常会导致与 DFT 计算的未校正数据相关的性能较差。实施了分子间相互作用的相对强度,以合理化 XCNSSN (X = F, Cl, Br, CN) 系列内的堆积趋势。这些相互作用的性质也通过分子中的原子方法进行了探索,该方法揭示了不同类型相互作用的分子间键合临界点,这些都是对 DTDA 自由基之间非共价相互作用的诊断。这些与之前对相关 DTDA 自由基的实验电荷密度研究进行了比较。然后将性能最佳的 DFT 方法应用于较大自由基的 α- 和 β-多晶型物 这些相互作用的性质也通过分子中的原子方法进行了探索,该方法揭示了不同类型相互作用的分子间键合临界点,这些都是对 DTDA 自由基之间非共价相互作用的诊断。这些与之前对相关 DTDA 自由基的实验电荷密度研究进行了比较。然后将性能最佳的 DFT 方法应用于较大自由基的 α- 和 β-多晶型物 这些相互作用的性质也通过分子中的原子方法进行了探索,该方法揭示了不同类型相互作用的分子间键合临界点,这些都是对 DTDA 自由基之间非共价相互作用的诊断。这些与之前对相关 DTDA 自由基的实验电荷密度研究进行了比较。然后将性能最佳的 DFT 方法应用于较大自由基的 α- 和 β-多晶型物p -NCC 6 F 4 CNSSN 对于基准研究来说太大了。这些证实了 α 和 β 相之间的微小能量差异,前五种表现最佳的方法和函数正确地反映了相对相稳定性。

























 京公网安备 11010802027423号
京公网安备 11010802027423号