当前位置:
X-MOL 学术
›
Acta Crystallogr. A Found. Adv.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
On the calculation of the electrostatic potential, electric field and electric field gradient from the aspherical pseudoatom model. II. Evaluation of the properties in an infinite crystal
Acta Crystallographica Section A: Foundations and Advances ( IF 1.8 ) Pub Date : 2021-07-29 , DOI: 10.1107/s2053273321005532 Jessie Weatherly 1 , Piero Macchi 2 , Anatoliy Volkov 1
Acta Crystallographica Section A: Foundations and Advances ( IF 1.8 ) Pub Date : 2021-07-29 , DOI: 10.1107/s2053273321005532 Jessie Weatherly 1 , Piero Macchi 2 , Anatoliy Volkov 1
Affiliation
|
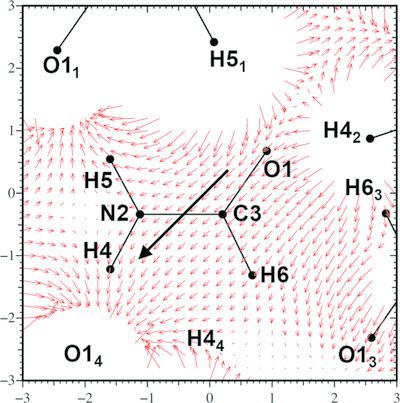
The previously reported exact potential and multipole moment (EP/MM) method for fast and precise evaluation of the intermolecular electrostatic interaction energies in molecular crystals using the pseudoatom representation of the electron density [Nguyen, Macchi & Volkov (2020), Acta Cryst. A76, 630–651] has been extended to the calculation of the electrostatic potential (ESP), electric field (EF) and electric field gradient (EFG) in an infinite crystal. The presented approach combines an efficient Ewald-type summation (ES) of atomic multipoles up to the hexadecapolar level in direct and reciprocal spaces with corrections for (i) the net polarization of the sample (the `surface term') due to a net dipole moment of the crystallographic unit cell (if present) and (ii) the short-range electron-density penetration effects. The rederived and reported closed-form expressions for all terms in the ES algorithm have been augmented by the expressions for the surface term available in the literature [Stenhammar, Trulsson & Linse (2011), J. Chem. Phys.134, 224104] and the exact potential expressions reported in a previous study [Volkov, King, Coppens & Farrugia (2006), Acta Cryst. A62, 400–408]. The resulting algorithm, coded using Fortran in the XDPROP module of the software package XD, was tested on several small molecular crystal systems (formamide, benzene, l-dopa, paracetamol, amino acids etc.) and compared with a series of EP/MM-based direct-space summations (DS) performed within a certain number of unit cells generated along both the positive and negative crystallographic directions. The EP/MM-based ES technique allows for a noticeably more precise determination of the EF and EFG and significantly better precision of the evaluated ESP when compared with the DS calculations, even when the latter include contributions from an array of symmetry-equivalent atoms generated within four additional unit cells along each crystallographic direction. In terms of computational performance, the ES/EP/MM method is significantly faster than the DS calculations performed within the extended unit-cell limits but trails the DS calculations within the reduced summation ranges. Nonetheless, the described EP/MM-based ES algorithm is superior to the direct-space summations as it does not require the user to monitor continuously the convergence of the evaluated properties as a function of the summation limits and offers a better precision–performance balance.
中文翻译:

从非球面伪原子模型计算静电势、电场和电场梯度。二、无限晶体的性质评估
先前报道的精确电位和多极矩 (EP/MM) 方法使用电子密度的赝原子表示快速精确地评估分子晶体中的分子间静电相互作用能 [Nguyen、Macchi & Volkov (2020),Acta Cryst。一个76, 630–651] 已扩展到无限晶体中静电势 (ESP)、电场 (EF) 和电场梯度 (EFG) 的计算。所提出的方法结合了在直接和倒易空间中高达十六极水平的原子多极的有效 Ewald 型求和 (ES) 与 (i) 由于净偶极子导致的样品净极化(“表面项”)的校正晶胞的矩(如果存在)和(ii)短程电子密度渗透效应。ES 算法中所有项的重新导出和报告的闭式表达式已通过文献中可用的表面项的表达式得到增强 [Stenhammar, Trulsson & Linse (2011), J. Chem. 物理。134, 224104] 以及先前研究 [Volkov, King, Coppens & Farrugia (2006), Acta Cryst. 中报道的确切潜在表达。A 62 , 400–408]。将所得的算法中,在使用的Fortran编码XDPROP软件包的模块XD,于几个小分子晶体系统(甲酰胺,苯,测试升-DOPA,扑热息痛,氨基酸等) 并与一系列基于 EP/MM 的直接空间求和 (DS) 进行比较,这些求和在一定数量的晶胞中沿正负晶体方向生成。与 DS 计算相比,基于 EP/MM 的 ES 技术可以显着更精确地确定 EF 和 EFG,并且评估 ESP 的精度显着提高,即使后者包括来自生成的对称等效原子阵列的贡献在沿每个结晶方向的四个附加晶胞内。在计算性能方面,ES/EP/MM 方法明显快于在扩展晶胞范围内执行的 DS 计算,但在缩减的求和范围内落后于 DS 计算。尽管如此,
更新日期:2021-09-02
中文翻译:
从非球面伪原子模型计算静电势、电场和电场梯度。二、无限晶体的性质评估
先前报道的精确电位和多极矩 (EP/MM) 方法使用电子密度的赝原子表示快速精确地评估分子晶体中的分子间静电相互作用能 [Nguyen、Macchi & Volkov (2020),Acta Cryst。一个76, 630–651] 已扩展到无限晶体中静电势 (ESP)、电场 (EF) 和电场梯度 (EFG) 的计算。所提出的方法结合了在直接和倒易空间中高达十六极水平的原子多极的有效 Ewald 型求和 (ES) 与 (i) 由于净偶极子导致的样品净极化(“表面项”)的校正晶胞的矩(如果存在)和(ii)短程电子密度渗透效应。ES 算法中所有项的重新导出和报告的闭式表达式已通过文献中可用的表面项的表达式得到增强 [Stenhammar, Trulsson & Linse (2011), J. Chem. 物理。134, 224104] 以及先前研究 [Volkov, King, Coppens & Farrugia (2006), Acta Cryst. 中报道的确切潜在表达。A 62 , 400–408]。将所得的算法中,在使用的Fortran编码XDPROP软件包的模块XD,于几个小分子晶体系统(甲酰胺,苯,测试升-DOPA,扑热息痛,氨基酸等) 并与一系列基于 EP/MM 的直接空间求和 (DS) 进行比较,这些求和在一定数量的晶胞中沿正负晶体方向生成。与 DS 计算相比,基于 EP/MM 的 ES 技术可以显着更精确地确定 EF 和 EFG,并且评估 ESP 的精度显着提高,即使后者包括来自生成的对称等效原子阵列的贡献在沿每个结晶方向的四个附加晶胞内。在计算性能方面,ES/EP/MM 方法明显快于在扩展晶胞范围内执行的 DS 计算,但在缩减的求和范围内落后于 DS 计算。尽管如此,

























 京公网安备 11010802027423号
京公网安备 11010802027423号