JACC: Heart Failure ( IF 13.0 ) Pub Date : 2021-07-26 , DOI: 10.1016/j.jchf.2021.05.019 Milton Packer 1
|
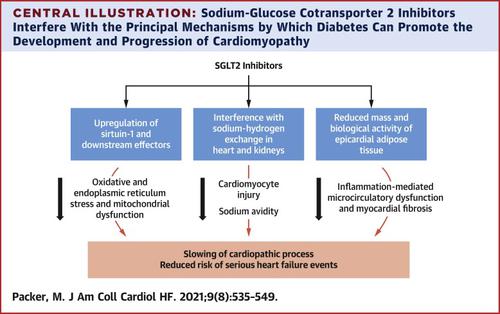
Diabetes promotes the development of both heart failure with a reduced ejection fraction and heart failure with a preserved ejection fraction through diverse mechanisms, which are likely mediated through hyperinsulinemia rather than hyperglycemia. Diabetes promotes nutrient surplus signaling (through Akt and mammalian target of rapamycin complex 1) and inhibits nutrient deprivation signaling (through sirtuin-1 and its downstream effectors); this suppresses autophagy and promotes endoplasmic reticulum and oxidative stress and mitochondrial dysfunction, thereby undermining the health of diabetic cardiomyocytes. The hyperinsulinemia of diabetes may also activate sodium-hydrogen exchangers in cardiomyocytes (leading to injury and loss) and in the proximal renal tubules (leading to sodium retention). Diabetes may cause epicardial adipose tissue expansion, and the resulting secretion of proinflammatory adipocytokines onto the adjoining myocardium can lead to coronary microcirculatory dysfunction and myocardial inflammation and fibrosis. Interestingly, sodium-glucose cotransporter 2 (SGLT2) inhibitors—the only class of antidiabetic medication that reduces serious heart failure events—may act to mitigate each of these mechanisms. SGLT2 inhibitors up-regulate sirtuin-1 and its downstream effectors and autophagic flux, thus explaining the actions of these drugs to reduce oxidative stress, normalize mitochondrial structure and function, and mute proinflammatory pathways in the stressed myocardium. Inhibition of SGLT2 may also lead to a reduction in the activity of sodium-hydrogen exchangers in the kidney (leading to diuresis) and in the heart (attenuating the development of cardiac hypertrophy and systolic dysfunction). Finally, SGLT2 inhibitors reduce the mass and mute the adverse biology of epicardial adipose tissue (and reduce the secretion of leptin), thus explaining the capacity of these drugs to mitigate myocardial inflammation, microcirculatory dysfunction, and fibrosis, and improve ventricular filling dynamics. The pathophysiological mechanisms by which SGLT2 inhibitors may benefit heart failure likely differ depending on ejection fraction, but each represents interference with distinct pathways by which hyperinsulinemia may adversely affect cardiac structure and function.
中文翻译:
糖尿病射血分数降低或保留的心力衰竭的不同病理生理机制
糖尿病通过多种机制促进射血分数降低的心力衰竭和射血分数保留的心力衰竭的发展,这可能是通过高胰岛素血症而不是高血糖介导的。糖尿病促进营养过剩信号(通过 Akt 和雷帕霉素复合物 1 的哺乳动物靶标)并抑制营养缺乏信号(通过 Sirtuin-1 及其下游效应器);这会抑制自噬并促进内质网和氧化应激和线粒体功能障碍,从而破坏糖尿病心肌细胞的健康。糖尿病的高胰岛素血症也可能激活心肌细胞(导致损伤和丢失)和近端肾小管(导致钠潴留)中的钠氢交换剂。糖尿病可引起心外膜脂肪组织扩张,由此产生的促炎性脂肪细胞因子分泌到相邻的心肌可导致冠状动脉微循环功能障碍和心肌炎症和纤维化。有趣的是,钠-葡萄糖协同转运蛋白 2 (SGLT2) 抑制剂——唯一一类减少严重心力衰竭事件的抗糖尿病药物——可能会减轻这些机制中的每一种。SGLT2 抑制剂上调 Sirtuin-1 及其下游效应子和自噬通量,从而解释了这些药物在减少氧化应激、使线粒体结构和功能正常化以及使受压心肌中的促炎通路静音的作用。SGLT2 的抑制也可能导致肾脏(导致利尿)和心脏(减弱心脏肥大和收缩功能障碍的发展)中钠-氢交换剂的活性降低。最后,SGLT2 抑制剂减少了心外膜脂肪组织的质量并消除了不良生物学(并减少了瘦素的分泌),从而解释了这些药物减轻心肌炎症、微循环功能障碍和纤维化以及改善心室充盈动态的能力。SGLT2 抑制剂可能有益于心力衰竭的病理生理机制可能因射血分数而异,但每种机制都代表了对高胰岛素血症可能对心脏结构和功能产生不利影响的不同途径的干扰。
























 京公网安备 11010802027423号
京公网安备 11010802027423号