Computers in Biology and Medicine ( IF 7.7 ) Pub Date : 2021-07-17 , DOI: 10.1016/j.compbiomed.2021.104656 Abdullah Ammar Karcioglu 1 , Hasan Bulut 1
|
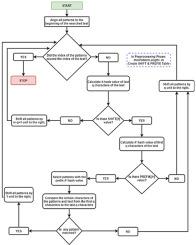
The string matching algorithms are among the essential fields in computer science, such as text search, intrusion detection systems, fraud detection, sequence search in bioinformatics. The exact string matching algorithms are divided into two parts: single and multiple. Multiple string matching algorithms involve finding elements of the pattern set P in a given input text T. String matching processes should be done in a time-efficient manner for DNA sequences. As the volume of the text T increases and the number of search patterns increases, the total runtime increases. Efficient algorithms should be selected to perform these search operations as soon as possible. In this study, the Wu-Manber algorithm, one of the multiple exact string matching algorithms, is improved. Although the Wu-Manber algorithm is effective, it has some limitations, such as hash collisions. In this study, the WM-q algorithm, a version of the Wu-Manber algorithm based on the perfect hash function for DNA sequences, is proposed. String matching is performed using different block lengths provided by the perfect hash function instead of using the fixed block length as in the traditional Wu-Manber algorithm. The proposed approach has been compared with E. Coli and Human Chromosome1 datasets, frequently used in the literature, using multiple exact string matching algorithms. The proposed algorithm gives better results for performance metrics such as the average runtime, the average number of characters and hash comparisons.
中文翻译:
DNA序列的WM-q多重精确串匹配算法
字符串匹配算法是计算机科学中的重要领域之一,例如文本搜索、入侵检测系统、欺诈检测、生物信息学中的序列搜索。精确的字符串匹配算法分为两个部分:单个和多个。多个字符串匹配算法涉及在给定的输入文本 T 中查找模式集 P 的元素。对于 DNA 序列,字符串匹配过程应该以节省时间的方式完成。随着文本 T 的量增加和搜索模式数量的增加,总运行时间增加。应该选择高效的算法来尽快执行这些搜索操作。本研究对多种字符串精确匹配算法之一的Wu-Manber算法进行了改进。Wu-Manber 算法虽然有效,但也有一定的局限性,比如哈希冲突。在这项研究中,提出了 WM-q 算法,它是基于 DNA 序列完美散列函数的 Wu-Manber 算法的一个版本。字符串匹配是使用完美散列函数提供的不同块长度来执行的,而不是像传统的 Wu-Manber 算法那样使用固定的块长度。所提出的方法已与文献中经常使用的大肠杆菌和人类染色体 1 数据集进行了比较,使用多个精确的字符串匹配算法。所提出的算法为性能指标提供了更好的结果,例如平均运行时间、平均字符数和哈希比较。字符串匹配是使用完美散列函数提供的不同块长度来执行的,而不是像传统的 Wu-Manber 算法那样使用固定的块长度。已将所提出的方法与文献中经常使用的大肠杆菌和人类染色体 1 数据集进行比较,使用多个精确的字符串匹配算法。所提出的算法为性能指标提供了更好的结果,例如平均运行时间、平均字符数和哈希比较。字符串匹配是使用完美散列函数提供的不同块长度来执行的,而不是像传统的 Wu-Manber 算法那样使用固定的块长度。所提出的方法已与文献中经常使用的大肠杆菌和人类染色体 1 数据集进行了比较,使用多个精确的字符串匹配算法。所提出的算法为性能指标提供了更好的结果,例如平均运行时间、平均字符数和哈希比较。

























 京公网安备 11010802027423号
京公网安备 11010802027423号