当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Asymmetric Synthesis of Fluoro, Fluoromethyl, Difluoromethyl, and Trifluoromethylcyclopropanes
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2021-07-07 , DOI: 10.1021/acs.accounts.1c00261 Amandine Pons 1 , Laetitia Delion 1, 2 , Thomas Poisson 1, 3 , André B Charette 2 , Philippe Jubault 1
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2021-07-07 , DOI: 10.1021/acs.accounts.1c00261 Amandine Pons 1 , Laetitia Delion 1, 2 , Thomas Poisson 1, 3 , André B Charette 2 , Philippe Jubault 1
Affiliation
|
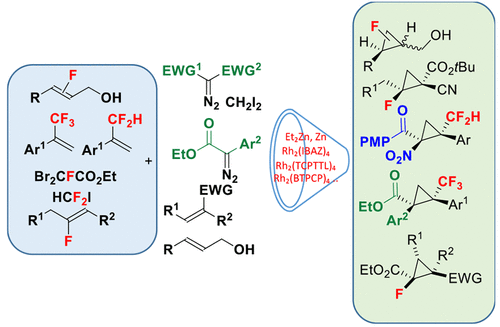
Fluorine-containing cyclopropanes are a subclass of cyclopropane derivatives that have generated considerable interest in medicinal chemistry for several decades. The replacement of a cyclopropane C–H or C–CH3 bond with fluorine or a fluorinated group (such as CF3 or CF2H) can lead sometimes to synergistic effects in terms of biological activity and improved metabolic profile of a cyclopropane containing bioactive compound. In this context, the preparation of fluoro-, difluoromethyl-, or trifluoromethyl-cyclopropane is particularly attractive and important but quite challenging considering the unique electronic properties that result from the incorporation of a fluorine atom into a substrate or a reagent. In the past decade, we have sought to develop new routes for the stereoselective synthesis of these building blocks using the most reliable cyclopropanation methods and convenient and readily available starting materials. The challenge that had to be undertaken was how we could use the unique properties of the fluorine atom to improve upon the efficiency of a given process rather than shutting it down. This could be overcome by defining new substrate/reagent reactivity guidelines and carefully selecting whether the fluorinated group was introduced on the electrophilic or nucleophilic partner for a given reaction. In this Account, we describe our contributions in this area that take advantage of diazo-derived rhodium carbenes, zinc carbenoids, ring closure processes, and biocatalytic methods to access these important potential drug subunits. Our initial investigation relied on the development of a Michael-initiated ring closure reaction using the Reformatsky enolate derived from readily available ethyl dibromofluoroacetate and α,β-unsaturated electrophiles. The reaction proceeded extremely well but with modest to good diastereoselectivities with ester acrylates. Further extension to various fluorinated nucleophiles such as oxazolidinone based and DABCO ylides led to similar selectivities.
中文翻译:

氟、氟甲基、二氟甲基和三氟甲基环丙烷的不对称合成
含氟环丙烷是环丙烷衍生物的一个子类,几十年来在药物化学中引起了极大的兴趣。替换环丙烷CH或C-CH的3键被氟或氟化基团(如CF 3或CF 2H) 有时可以在生物活性和改善的含环丙烷生物活性化合物的代谢特征方面产生协同效应。在这种情况下,氟-、二氟甲基-或三氟甲基-环丙烷的制备是特别有吸引力和重要的,但考虑到由于将氟原子结合到底物或试剂中而产生的独特电子特性,因此非常具有挑战性。在过去的十年中,我们一直在寻求使用最可靠的环丙烷化方法和方便且容易获得的起始材料来开发立体选择性合成这些结构单元的新路线。必须承担的挑战是我们如何利用氟原子的独特特性来提高给定过程的效率,而不是将其关闭。这可以通过定义新的底物/试剂反应性指南并仔细选择氟化基团是在给定反应的亲电子或亲核伙伴上引入来克服。在这个帐户中,我们描述了我们在该领域的贡献,这些贡献利用重氮衍生的铑卡宾、锌卡宾、闭环过程和生物催化方法来获取这些重要的潜在药物亚基。我们的初步研究依赖于使用衍生自现成的二溴氟乙酸乙酯和 α,β-不饱和亲电子试剂的 Reformatsky 烯醇盐开发迈克尔引发的闭环反应。反应进行得非常好,但与丙烯酸酯的非对映选择性适中到良好。
更新日期:2021-07-20
中文翻译:
氟、氟甲基、二氟甲基和三氟甲基环丙烷的不对称合成
含氟环丙烷是环丙烷衍生物的一个子类,几十年来在药物化学中引起了极大的兴趣。替换环丙烷CH或C-CH的3键被氟或氟化基团(如CF 3或CF 2H) 有时可以在生物活性和改善的含环丙烷生物活性化合物的代谢特征方面产生协同效应。在这种情况下,氟-、二氟甲基-或三氟甲基-环丙烷的制备是特别有吸引力和重要的,但考虑到由于将氟原子结合到底物或试剂中而产生的独特电子特性,因此非常具有挑战性。在过去的十年中,我们一直在寻求使用最可靠的环丙烷化方法和方便且容易获得的起始材料来开发立体选择性合成这些结构单元的新路线。必须承担的挑战是我们如何利用氟原子的独特特性来提高给定过程的效率,而不是将其关闭。这可以通过定义新的底物/试剂反应性指南并仔细选择氟化基团是在给定反应的亲电子或亲核伙伴上引入来克服。在这个帐户中,我们描述了我们在该领域的贡献,这些贡献利用重氮衍生的铑卡宾、锌卡宾、闭环过程和生物催化方法来获取这些重要的潜在药物亚基。我们的初步研究依赖于使用衍生自现成的二溴氟乙酸乙酯和 α,β-不饱和亲电子试剂的 Reformatsky 烯醇盐开发迈克尔引发的闭环反应。反应进行得非常好,但与丙烯酸酯的非对映选择性适中到良好。


























 京公网安备 11010802027423号
京公网安备 11010802027423号