当前位置:
X-MOL 学术
›
J. Heterocycl. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Synthesis of non-toxic anticancer active forskolin-indole-triazole conjugates along with their in silico succinate dehydrogenase inhibition studies
Journal of Heterocyclic Chemistry ( IF 2.4 ) Pub Date : 2021-06-27 , DOI: 10.1002/jhet.4332 Ponnam Devendar 1 , A. Niranjana Kumar 1 , K. V. N. Satya Srinivas 1 , J. Kotesh Kumar 1 , Shilpi Singh 2 , Abha Meena 3 , Pallavi Misra 2 , Suaib Luqman 2
Journal of Heterocyclic Chemistry ( IF 2.4 ) Pub Date : 2021-06-27 , DOI: 10.1002/jhet.4332 Ponnam Devendar 1 , A. Niranjana Kumar 1 , K. V. N. Satya Srinivas 1 , J. Kotesh Kumar 1 , Shilpi Singh 2 , Abha Meena 3 , Pallavi Misra 2 , Suaib Luqman 2
Affiliation
|
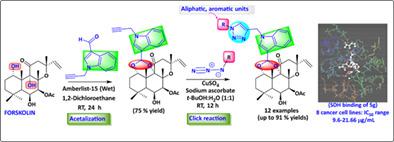
Biologically important three different pharmacophores, forskolin, indole and 1,2,3-triazoles are coupled to obtain a hybrid molecule. Here, we described the synthesis of novel series of forskolin-indole-triazole conjugates 5a-5l by using the Cu(I) catalyzed 1,3-dipolar cycloaddition reaction. Furthermore, the biological significance of the synthesized molecules was assessed by in silico and in vitro modes. All the synthesized compounds were evaluated for in vitro anticancer activity against PC-3, MCF-7, MDA-MB-231, COLO-205, HeLa, WRL-68, RAJI, CHANG and RAW-264.7 cell lines. Compound 5g was found to be the most potent in all the tested cell lines (IC50 range 9.6–21.66 μg/ml, except COLO-205), 5a, 5b and 5k were observed to exert its effect only against WRL-68 (IC50 range 27.69–48.18 μg/ml), when compared to parent 3 (IC50 > 100 μg/ml, tested concentrations 10–50 μg/ml) and standard Doxorubicin (IC50 range 0.42–3.16 μg/ml). The most potent compound 5g (MEF50 0.57) was found non-toxic to human erythrocytes as compared to control (MEF50 0.60) at tested concentration (50 μg/ml). In silico-based succinate dehydrogenase inhibition showed that the synthesized compounds were having potent binding affinity compared to forskolin. Predictive ADMET and toxicity risk assessment analysis revealed that most of the compounds were complying with the standard limit of Lipinski's rule of five for oral bioavailability.
中文翻译:

无毒抗癌活性毛喉素-吲哚-三唑偶联物的合成及其在硅片上的琥珀酸脱氢酶抑制研究
生物学上重要的三种不同的药效团,毛喉素、吲哚和 1,2,3-三唑偶联以获得混合分子。在这里,我们描述了使用 Cu(I) 催化的 1,3-偶极环加成反应合成新系列的毛喉素-吲哚-三唑共轭物 5a-5l。此外,合成分子的生物学意义通过计算机模拟和体外模式进行评估。评估了所有合成化合物对 PC-3、MCF-7、MDA-MB-231、COLO-205、HeLa、WRL-68、RAJI、CHANG 和 RAW-264.7 细胞系的体外抗癌活性。发现化合物 5g 在所有测试的细胞系中最有效(IC 50范围为 9.6–21.66 μg/ml,COLO-205 除外),观察到 5a、5b 和 5k 仅对 WRL-68 发挥作用(IC 50范围 27.69–48.18 μg/ml),与亲本 3(IC 50 > 100 μg/ml,测试浓度 10–50 μg/ml)和标准多柔比星(IC 50范围 0.42–3.16 μg/ml)相比。与对照 (MEF 50 0.60)相比,在测试浓度 (50 μg/ml)下发现最有效的化合物 5g (MEF 50 0.57) 对人红细胞无毒。基于硅的琥珀酸脱氢酶抑制表明,与毛喉素相比,合成的化合物具有强大的结合亲和力。预测性 ADMET 和毒性风险评估分析表明,大多数化合物符合 Lipinski 的口服生物利用度五法则的标准限值。
更新日期:2021-06-27
中文翻译:
无毒抗癌活性毛喉素-吲哚-三唑偶联物的合成及其在硅片上的琥珀酸脱氢酶抑制研究
生物学上重要的三种不同的药效团,毛喉素、吲哚和 1,2,3-三唑偶联以获得混合分子。在这里,我们描述了使用 Cu(I) 催化的 1,3-偶极环加成反应合成新系列的毛喉素-吲哚-三唑共轭物 5a-5l。此外,合成分子的生物学意义通过计算机模拟和体外模式进行评估。评估了所有合成化合物对 PC-3、MCF-7、MDA-MB-231、COLO-205、HeLa、WRL-68、RAJI、CHANG 和 RAW-264.7 细胞系的体外抗癌活性。发现化合物 5g 在所有测试的细胞系中最有效(IC 50范围为 9.6–21.66 μg/ml,COLO-205 除外),观察到 5a、5b 和 5k 仅对 WRL-68 发挥作用(IC 50范围 27.69–48.18 μg/ml),与亲本 3(IC 50 > 100 μg/ml,测试浓度 10–50 μg/ml)和标准多柔比星(IC 50范围 0.42–3.16 μg/ml)相比。与对照 (MEF 50 0.60)相比,在测试浓度 (50 μg/ml)下发现最有效的化合物 5g (MEF 50 0.57) 对人红细胞无毒。基于硅的琥珀酸脱氢酶抑制表明,与毛喉素相比,合成的化合物具有强大的结合亲和力。预测性 ADMET 和毒性风险评估分析表明,大多数化合物符合 Lipinski 的口服生物利用度五法则的标准限值。


























 京公网安备 11010802027423号
京公网安备 11010802027423号