Molecular Diversity ( IF 3.8 ) Pub Date : 2021-06-19 , DOI: 10.1007/s11030-021-10250-2 Chiakang Hung 1 , Giuseppina Gini 1
|
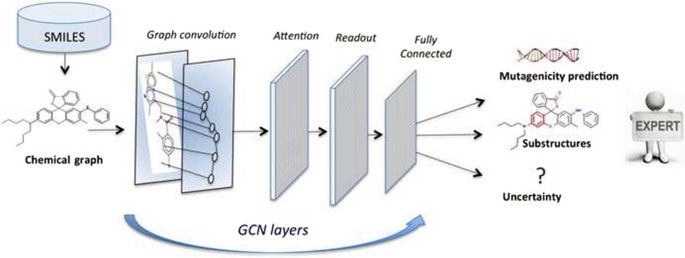
Deep neural networks are effective in learning directly from low-level encoded data without the need of feature extraction. This paper shows how QSAR models can be constructed from 2D molecular graphs without computing chemical descriptors. Two graph convolutional neural network-based models are presented with and without a Bayesian estimation of the prediction uncertainty. The property under investigation is mutagenicity: Models developed here predict the output of the Ames test. These models take the SMILES representation of the molecules as input to produce molecular graphs in terms of adjacency matrices and subsequently use attention mechanisms to weight the role of their subgraphs in producing the output. The results positively compare with current state-of-the-art models. Furthermore, our proposed model interpretation can be enhanced by the automatic extraction of the substructures most important in driving the prediction, as well as by uncertainty estimations.
Graphic abstract
中文翻译:
使用图卷积神经网络进行无描述符 QSAR 建模:致突变性预测的案例
深度神经网络可以有效地直接从低级编码数据中学习,而无需进行特征提取。本文展示了如何在不计算化学描述符的情况下从 2D 分子图构建 QSAR 模型。提出了两个基于图卷积神经网络的模型,有和没有预测不确定性的贝叶斯估计。正在研究的特性是致突变性:这里开发的模型可以预测 Ames 测试的结果。这些模型将分子的 SMILES 表示作为输入,根据邻接矩阵生成分子图,然后使用注意机制来衡量其子图在生成输出中的作用。结果与当前最先进的模型相比较。此外,
























 京公网安备 11010802027423号
京公网安备 11010802027423号