Journal of the Taiwan Institute of Chemical Engineers ( IF 5.7 ) Pub Date : 2021-06-11 , DOI: 10.1016/j.jtice.2021.05.048 Afsaneh Ghahari , Heidar Raissi , Farzaneh Farzad
|
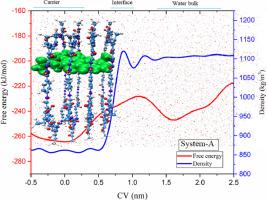
The adsorption mechanism of doxorubicin (DOX) on pristine covalent organic frameworks (COF) and functionalized COF (F-COF) is investigated by using molecular dynamics (MD) and Metadynamics simulations. MD simulations are performed to explore the loading of DOX into the COFs. To evaluate this process, a set of descriptors, including interaction energies, radial distribution function, and square mean displacement, the number of hydrogen bonds (HB) are calculated throughout the simulation trajectories. It is determined that HB interactions are the most important factors in stability of the investigated systems. The MD results show that the drug molecules move toward the COFs and form the stable complexes. Functionalization of COF with six and three hydroxyl groups increases its interaction energy with DOX from -16.87 to -264.95 and -139.87 kJ/mol, respectively. In this study, the free energy surface (FES) is also calculated by well-tempered metadynamics simulation technique. The PES landscapes confirm that the global minimum could be typically relevant to formation of the HB. The free energy values for the COF/DOX complexes in pristine and functionalization of COFs at their global minima are reached about -229.7, -260.46 (for six OH groups), and -243.37 (for three OH groups) kJ/mol, respectively. In addition, at the acidic condition, protonation of DOX causes that the interactions between DOXs and the COFs become weaker and drug molecules could release from the nanocarrier cavities.
中文翻译:
基于表面功能化二维共价有机骨架的新型给药平台设计
使用分子动力学 (MD) 和元动力学模拟研究了阿霉素 (DOX) 在原始共价有机框架 (COF) 和功能化 COF (F-COF) 上的吸附机制。执行 MD 模拟以探索 DOX 加载到 COF 中。为了评估这个过程,在整个模拟轨迹中计算了一组描述符,包括相互作用能、径向分布函数和均方位移,氢键 (HB) 的数量。确定 HB 相互作用是研究系统稳定性的最重要因素。MD 结果表明药物分子向 COF 移动并形成稳定的复合物。具有六个和三个羟基的 COF 官能化将其与 DOX 的相互作用能从 -16.87 增加到 -264.95 和 -139.87 kJ/mol,分别。在这项研究中,自由能面 (FES) 也是通过良好的元动力学模拟技术计算的。PES 景观确认全球最小值通常与 HB 的形成有关。COF/DOX 复合物的原始和 COF 功能化在其全局最小值时的自由能值分别达到约 -229.7、-260.46(对于六个 OH 基团)和 -243.37(对于三个 OH 基团)kJ/mol。此外,在酸性条件下,DOX 的质子化导致 DOX 与 COF 之间的相互作用变弱,药物分子可能会从纳米载体腔中释放出来。PES 景观确认全球最小值通常与 HB 的形成有关。COF/DOX 复合物的原始和 COF 功能化在其全局最小值时的自由能值分别达到约 -229.7、-260.46(对于六个 OH 基团)和 -243.37(对于三个 OH 基团)kJ/mol。此外,在酸性条件下,DOX 的质子化导致 DOX 与 COF 之间的相互作用变弱,药物分子可能会从纳米载体腔中释放出来。PES 景观确认全球最小值通常与 HB 的形成有关。COF/DOX 复合物的原始和 COF 功能化在其全局最小值时的自由能值分别达到约 -229.7、-260.46(对于六个 OH 基团)和 -243.37(对于三个 OH 基团)kJ/mol。此外,在酸性条件下,DOX 的质子化导致 DOX 与 COF 之间的相互作用变弱,药物分子可能会从纳米载体腔中释放出来。
























 京公网安备 11010802027423号
京公网安备 11010802027423号