当前位置:
X-MOL 学术
›
Acta Cryst. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Revisiting stacking interactions in tetrathiafulvalene and selected derivatives using tight-binding quantum chemical calculations and local coupled-cluster method
Acta Crystallographica Section B ( IF 2.684 ) Pub Date : 2021-05-13 , DOI: 10.1107/s2052520621003085 Kang Zheng , Danping Li , Liu Jiang , Xiaowei Li , Changjian Xie , Ling Feng , Jie Qin , Shaosong Qian , Qiuxiang Pang
Acta Crystallographica Section B ( IF 2.684 ) Pub Date : 2021-05-13 , DOI: 10.1107/s2052520621003085 Kang Zheng , Danping Li , Liu Jiang , Xiaowei Li , Changjian Xie , Ling Feng , Jie Qin , Shaosong Qian , Qiuxiang Pang
|
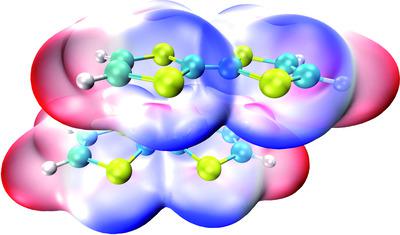
The engineering of supramolecular architectures needs accurate descriptions of the intermolecular interactions in crystal structures. Tetrathiafulvalene (TTF) is an effective building block used in the construction of promising functional materials. The parallel packing of the neutral TTF–TTF system was studied previously using the high-level quantum chemical method, advancing it as a valuable model system. The recently developed tight-binding quantum chemical method GFN2-xTB and local coupled-cluster method DLPNO-CCSD(T) were used to investigate the stacking interactions of TTF and selected derivatives deposited in the Cambridge Structural Database. Using the interaction energy of the TTF–TTF dimer calculated at the CCSD(T)/CBS level as the reference, the accuracies of the two methods are investigated. The energy decomposition analysis within the DLPNO-CCSD(T) framework reveals the importance of dispersion interaction in the TTF-related stacking systems. The dispersion interaction density plot vividly shows the magnitude and distribution of the dispersion interaction, providing a revealing insight into the stacking interactions in crystal structures. The results show that the GFN2-xTB and DLPNO-CCSD(T) methods could achieve accuracy at an affordable computational cost, which would be valuable in understanding the nature of parallel stacking in supramolecular systems.
中文翻译:

使用紧束缚量子化学计算和局部耦合簇方法重新审视四硫富瓦烯和选定衍生物中的堆积相互作用
超分子结构的工程需要准确描述晶体结构中的分子间相互作用。四硫富瓦烯 (TTF) 是一种有效的构建块,用于构建有前景的功能材料。先前使用高级量子化学方法研究了中性 TTF-TTF 系统的平行堆积,将其推进为有价值的模型系统。最近开发的紧束缚量子化学方法 GFN2-xTB 和局部耦合簇方法 DLPNO-CCSD(T) 用于研究 TTF 和剑桥结构数据库中存放的选定衍生物的堆叠相互作用。使用在 CCSD(T)/CBS 水平计算的 TTF-TTF 二聚体的相互作用能作为参考,研究了两种方法的准确性。DLPNO-CCSD(T) 框架内的能量分解分析揭示了色散相互作用在 TTF 相关堆叠系统中的重要性。色散相互作用密度图生动地显示了色散相互作用的大小和分布,提供了对晶体结构中堆叠相互作用的揭示。结果表明,GFN2-xTB 和 DLPNO-CCSD(T) 方法可以以可承受的计算成本实现准确性,这对于理解超分子系统中并行堆叠的性质非常有价值。提供对晶体结构中堆叠相互作用的揭示性洞察。结果表明,GFN2-xTB 和 DLPNO-CCSD(T) 方法可以以可承受的计算成本实现准确性,这对于理解超分子系统中并行堆叠的性质非常有价值。提供对晶体结构中堆叠相互作用的揭示性洞察。结果表明,GFN2-xTB 和 DLPNO-CCSD(T) 方法可以以可承受的计算成本实现准确性,这对于理解超分子系统中并行堆叠的性质非常有价值。
更新日期:2021-06-07
中文翻译:
使用紧束缚量子化学计算和局部耦合簇方法重新审视四硫富瓦烯和选定衍生物中的堆积相互作用
超分子结构的工程需要准确描述晶体结构中的分子间相互作用。四硫富瓦烯 (TTF) 是一种有效的构建块,用于构建有前景的功能材料。先前使用高级量子化学方法研究了中性 TTF-TTF 系统的平行堆积,将其推进为有价值的模型系统。最近开发的紧束缚量子化学方法 GFN2-xTB 和局部耦合簇方法 DLPNO-CCSD(T) 用于研究 TTF 和剑桥结构数据库中存放的选定衍生物的堆叠相互作用。使用在 CCSD(T)/CBS 水平计算的 TTF-TTF 二聚体的相互作用能作为参考,研究了两种方法的准确性。DLPNO-CCSD(T) 框架内的能量分解分析揭示了色散相互作用在 TTF 相关堆叠系统中的重要性。色散相互作用密度图生动地显示了色散相互作用的大小和分布,提供了对晶体结构中堆叠相互作用的揭示。结果表明,GFN2-xTB 和 DLPNO-CCSD(T) 方法可以以可承受的计算成本实现准确性,这对于理解超分子系统中并行堆叠的性质非常有价值。提供对晶体结构中堆叠相互作用的揭示性洞察。结果表明,GFN2-xTB 和 DLPNO-CCSD(T) 方法可以以可承受的计算成本实现准确性,这对于理解超分子系统中并行堆叠的性质非常有价值。提供对晶体结构中堆叠相互作用的揭示性洞察。结果表明,GFN2-xTB 和 DLPNO-CCSD(T) 方法可以以可承受的计算成本实现准确性,这对于理解超分子系统中并行堆叠的性质非常有价值。


























 京公网安备 11010802027423号
京公网安备 11010802027423号