Applied Surface Science ( IF 6.7 ) Pub Date : 2021-04-30 , DOI: 10.1016/j.apsusc.2021.149822 Halima Said , Michal Novotný , Ivan Černušák , Tomáš Bučko
|
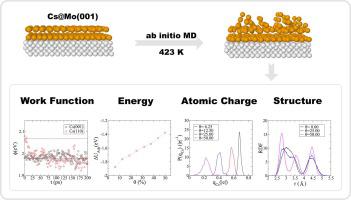
Ab initio molecular dynamics simulations have been conducted to investigate the properties of cesiated surface Mo(0 0 1) at the typical operating temperature of the negative hydrogen ion sources (423 K). In the systems with sub-monolayer Cs coverages (), both the mobility and ionicity of the adatoms decrease with . The computed shape of the work function () dependence on agrees with experiment, although our simulations underestimate the extent of the variation. In multilayer depositions, cesium forms two phases exhibiting distinctly different properties. The atoms laid directly on the substrate form an ordered layer with the atomic density and charges similar to those of the saturated monolayer (ML). A disordered phase with a much lower particle density is formed by the atoms stacked on the ordered layer and induces a positive shift in by 0.32 eV compared to ML. A similar behavior is observed for the Cs(0 0 1) and Cs(1 1 0) surfaces used as models for thick Cs depositions. The work function determined for multilayer Cs depositions at T = 423 K is nearly independent of the substrate face and of the presence/absence of Mo, indicating that of such systems is fully determined by the disordered surface phase.
中文翻译:
Cs在Mo(0 0 1)上吸附的从头算分子动力学研究:超越单层覆盖
从头开始进行分子动力学模拟,以研究在负氢离子源(423 K)的典型工作温度下,铯表面Mo(0 0 1)的性质。在具有亚单层Cs覆盖率的系统中(),原子的迁移率和离子性都会随着 。功函数的计算形状()依赖 同意实验,尽管我们的模拟低估了 变化。在多层沉积中,铯形成两相,表现出明显不同的性质。直接放置在基板上的原子形成具有原子密度且电荷类似于饱和单层(ML)的有序层。堆积在有序层上的原子形成了具有低得多的粒子密度的无序相,并引起了正向迁移。与ML相比降低了0.32 eV。对于用作厚Cs沉积模型的Cs(0 0 1)和Cs(1 1 0)表面,观察到了类似的行为。在T = 423 K时确定的多层Cs沉积的功函数几乎与衬底面以及是否存在Mo无关,这表明 这些系统的完全取决于无序的表面相。


























 京公网安备 11010802027423号
京公网安备 11010802027423号