Letters in Drug Design & Discovery ( IF 1 ) Pub Date : 2021-01-31 , DOI: 10.2174/1570180817999200905092444 Vijay Kumar Patel 1 , Harish Rajak 1
|
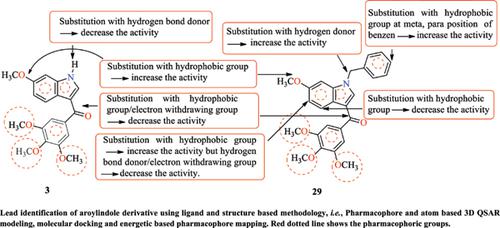
Background: The ligand and structure based integrated strategies are being repeatedly and effectively employed for the precise search and design of novel ligands against various disease targets. Aroylindole derivative has a similar structural analogy as Combretastatin A-4, and exhibited potent anticancer activity on several cancer cell lines.
Objective: To identify structural features of aroylindole derivatives through 3D-QSAR and multiple pharmacophore modelling for the search of novel colchicines inhibitor via virtual screening.
Methods: The present study utilizes ligand and structure based methodology for the establishment of structure activity correlation among trimethoxyaroylindole derivatives and the search of novel colchicines inhibitor via virtual screening. The 3D-QSAR studies were performed using Phase module and provided details of relationship between structure and biological activity. A single ligand based pharmacophore model was generated from Phase on compound 3 and compound 29 and three energetically optimized structure based pharmacophore models were generated from epharmacophore for co-crystallized ligand, compound 3 and compound 29 with protein PBD ID 1SA0, 5EYP and 5LYJ. These pharmacophoric features containing hit-like compounds were collected from commercially available ZINC database and screened using virtual screening workflow.
Results and Discussion: The 3D-QSAR model studies with good PLSs statistics for factor four was characterized by the best prediction coefficient Q2 (0.8122), regression R2 (0.9405), SD (0.2581), F (102.7), P (1.56e-015), RMSE (0.402), Stability (0.5411) and Pearson-r (0.9397). The generated epharmacophores have GH scores over 0.5 and AUAC ≥ 0.7 indicated that all the pharmacophores were suitable for pharmacophore-based virtual screening. The virtual screened compounds ZINC12323179, ZINC01642724, and ZINC14238006 have showed similar structural alignment as co-crystallized ligand and showed the hydrogen bonding of ligand with ASN101, SER178, THR179, VAL238, CYS241 amino acid of protein.
Conclusion: The study illustrates that the ligand and structure based pharmacophoric approach is beneficial for identification of structurally diverse hits, having better binding affinity on colchicines binding site as novel anticancer agents.
中文翻译:
通过3D-QSAR和多种药理学模型对芳基吲哚衍生物的结构研究,以寻找新型秋水仙碱抑制剂
背景:基于配体和结构的整合策略被反复有效地用于针对各种疾病靶标的新型配体的精确搜索和设计。芳酰基吲哚衍生物具有与Combretastatin A-4类似的结构类似物,并且在几种癌细胞系中表现出有效的抗癌活性。
目的:通过3D-QSAR和多种药效团模型鉴定芳基吲哚衍生物的结构特征,以通过虚拟筛选寻找新型秋水仙碱抑制剂。
方法:本研究利用基于配体和结构的方法建立三甲氧基芳基吲哚衍生物之间的结构活性相关性,并通过虚拟筛选寻找新型秋水仙碱抑制剂。使用相模块进行了3D-QSAR研究,并提供了结构与生物活性之间关系的详细信息。从化合物3和化合物29上的相生成了一个基于配体的单一药效团模型,并从电子药效团中生成了三种基于能量优化结构的药效团模型,用于共结晶配体,化合物3和化合物29以及蛋白PBD ID 1SA0、5EYP和5LYJ。从市售的ZINC数据库中收集包含命中样化合物的这些药效学特征,并使用虚拟筛选工作流程进行筛选。
结果与讨论:具有良好的PLS统计数据的4D因子3D-QSAR模型研究具有最佳预测系数Q 2(0.8122),回归R 2(0.9405),SD(0.2581),F(102.7),P(1.56) e-015),RMSE(0.402),稳定性(0.5411)和Pearson-r(0.9397)。产生的电子药团的GH分数超过0.5,并且AUAC≥0.7表明所有这些药团均适用于基于药效团的虚拟筛选。经过虚拟筛选的化合物ZINC12323179,ZINC01642724和ZINC14238006具有与共结晶的配体相似的结构排列,并显示该配体与蛋白质的ASN101,SER178,THR179,VAL238,CYS241氨基酸形成氢键。
结论:该研究表明基于配体和结构的药效学方法对于鉴定结构多样的命中物是有益的,作为新型抗癌剂对秋水仙碱的结合位点具有更好的结合亲和力。

























 京公网安备 11010802027423号
京公网安备 11010802027423号